
Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation
- 1Howard Hughes Medical Institute,
- 2Department of Biochemistry and Biophysics, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA;
- 3Cardiovascular Research Center, Massachusetts General Hospital, Boston, Massachusetts 02114, USA;
- 4Department of Stem Cell and Regenerative Biology, Harvard University, Cambridge, Massachusetts 02138, USA
Abstract
Ten-eleven translocation 1–3 (Tet1–3) proteins have recently been discovered in mammalian cells to be members of a family of DNA hydroxylases that possess enzymatic activity toward the methyl mark on the 5-position of cytosine (5-methylcytosine [5mC]), a well-characterized epigenetic modification that has essential roles in regulating gene expression and maintaining cellular identity. Tet proteins can convert 5mC into 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) through three consecutive oxidation reactions. These modified bases may represent new epigenetic states in genomic DNA or intermediates in the process of DNA demethylation. Emerging biochemical, genetic, and functional evidence suggests that Tet proteins are crucial for diverse biological processes, including zygotic epigenetic reprogramming, pluripotent stem cell differentiation, hematopoiesis, and development of leukemia. Insights into how Tet proteins contribute to dynamic changes in DNA methylation and gene expression will greatly enhance our understanding of epigenetic regulation of normal development and human diseases.
Keywords
The development of a multicellular organism follows a series of spatiotemporally defined biological processes. During development, stem and progenitor cells differentiate into functionally diverse cell types with unique gene expression profiles, which are regulated, at least in part, by cell type-specific covalent modifications of DNA and histones (Jaenisch and Bird 2003). These epigenetic modifications are largely responsible for cells with identical genomes stably displaying distinct cellular phenotypes.
DNA cytosine methylation is one of the best-characterized epigenetic modifications and plays important roles in a variety of cellular processes, including retrotransposon silencing, genomic imprinting, X-chromosome inactivation, regulation of gene expression, and maintenance of epigenetic memory (Bird 2002). In mammalian genomes, methylation of cytosine predominantly occurs at CpG dinucleotides; however, pervasive DNA methylation in non-CG contexts (CpH, where H = A, C, or T) has been reported in mouse and human embryonic stem (ES) cells (∼25% of all methylcytosines in human ES cells) (Ramsahoye et al. 2000; Lister et al. 2009). New DNA methylation patterns are initially established by de novo DNA methyltransferases: Dnmt3a and Dnmt3b (Okano et al. 1998; Okano et al. 1999). The pattern of DNA methylation is then faithfully maintained during DNA replication by the maintenance methyltransferase Dnmt1, which is recruited to replication foci via its physical interaction with the ubiquitin-like plant homeodomain and RING finger domain 1 (Uhrf1) that strongly binds to hemimethylated DNA (Bestor et al. 1988; Hermann et al. 2004; Bostick et al. 2007; Sharif et al. 2007). Deletion of Dnmt1 or Dnmt3b results in embryonic lethality, whereas homozygous Dnmt3a knockout mice die ∼4 wk after birth (Li et al. 1992; Okano et al. 1999), suggesting that DNA methyltransferases are essential for normal mammalian development. Mutations in the DNA methylation machinery have long been linked to inherited human diseases. For example, mutations in the human DNMT3B gene cause immunodeficiency centromeric instability facial anomalies (ICF) syndrome (Okano et al. 1999; Xu et al. 1999). Aberrant DNA methylation is also linked to imprinting disorders, such as Prader-Willi/Angelman syndrome (PWS/AS), as well as human cancers (Robertson 2005). Together, these studies have firmly established the importance of precise regulation of DNA methylation patterns in normal mammalian development.
Compared with readily reversible modifications of histone proteins, DNA methylation was generally considered to be a relatively stable epigenetic modification. However, bisulphite sequencing analysis as well as immunofluorescence staining using 5-methylcytosine (5mC)-specific antibodies indicated that global erasure of DNA methylation can take place in specific embryonic stages, such as zygotes (Mayer et al. 2000; Oswald et al. 2000) and developing primordial germ cells (PGCs) (Hajkova et al. 2002; Sasaki and Matsui 2008). In addition, recent genome-wide analysis of the DNA methylation pattern in pluripotent and differentiated cells at single-nucleotide resolution indicated that DNA methylation can be dynamically regulated during cellular differentiation (Meissner et al. 2008; Lister et al. 2009). These observations suggest the existence of mammalian enzymatic activities capable of erasing or modifying pre-existing DNA methylation patterns. However, the identity of such enzymes has been enigmatic and is the subject of intense research (Ooi and Bestor 2008; Wu and Zhang 2010).
Human ten-eleven translocation 1 (TET1) and mouse Tet proteins have recently been identified to have the capacity to convert 5mC to 5hmC (5-hydroxymethylcytosine) (Tahiliani et al. 2009; Ito et al. 2010), raising the possibility that 5mC distribution can be dynamically regulated by the Tet family of DNA hydroxylases. Furthermore, the presence of novel 5mC oxidation derivatives in genomic DNA may provide an additional layer of epigenetic information. In this review, we discuss the current knowledge about the mechanism and functions of Tet protein-mediated oxidation of 5mC. We begin by describing the recent progress toward elucidating the enzymatic activity of these DNA hydroxylases and detection methods for 5mC oxidation derivatives. We then discuss the role of Tet proteins in DNA demethylation as well as their roles in normal development and initiation of leukemia. We conclude by highlighting the remaining important questions in this emerging field.
Enzymatic activity of Tet proteins
The founding member of the Tet family of DNA hydroxylases, the TET1 gene, was initially identified in acute myeloid leukemia (AML) as a fusion partner of the histone H3 Lys 4 (H3K4) methyltransferase MLL (mixed-lineage leukemia) (Ono et al. 2002; Lorsbach et al. 2003). Rao and colleagues (Tahiliani et al. 2009) have recently shown that human TET1 protein possesses enzymatic activity capable of hydroxylating 5mC to generate 5hmC. They were interested in Tet proteins because of their sequence similarity to the Trypanosome base J (β-D-glucosyl-hydroxymethyl-uracil)-binding proteins JBP1 and JBP2 (Iyer et al. 2009), which are capable of hydrolyzing the methyl group of thymine (Borst and Sabatini 2008). Our group extended their finding by demonstrating that all members of the mouse Tet protein family (Tet1–3) have 5mC hydroxylase activities (Ito et al. 2010). Tet proteins contain several conserved domains (Fig. 1; Tahiliani et al. 2009), including a CXXC domain that has high affinity for clustered unmethylated CpG dinucleotides and a catalytic domain that is typical of Fe(II)- and 2-oxoglutarate (2OG)-dependent dioxygenases. In agreement with the known reaction mechanism of dioxygenases (Loenarz and Schofield 2011), mutation of putative iron-binding sites of Tet proteins abolishes their enzymatic activities (Tahiliani et al. 2009; Ito et al. 2010). In addition, 2-hydroxyglutarate (2-HG), a competitive inhibitor of 2OG-dependent dioxygenases, suppresses the catalytic activity of Tet proteins (W Xu et al. 2011). Interestingly, both fully methylated and hemimethylated DNA in a CG or non-CG context can serve as substrates for TET1 (Tahiliani et al. 2009; Ficz et al. 2011; Pastor et al. 2011).

Domain architecture of mouse Tet proteins. Schematic diagrams of predicted functional domains in the mouse Tet proteins (Tet1–3). Three conserved domains—including CXXC zinc finger, the cysteine-rich region (Cys-rich), and the double-stranded β-helix (DSBH) fold of the 2OG-Fe(II) dioxygenase domain—are indicated. Numbers represent the amino acid numbers. Note that Tet2 does not contain a predicted CXXC domain. Multiple sequence alignment of the catalytic motif of the JBP/Tet family of dioxygenases. Sequences used in the alignment include the Trypanosoma brucei JBP1 (Q9U6M3) and JBP2 (Q57X81); human TET1 (Q8NFU7), TET2 (Q6N021), and TET3 (O43151); and mouse Tet1 (GU079948), Tet2 (GU079949), and Tet3 (Q8BG87). Numbers represent the amino acid numbers. Alignment was performed using MultAlin (http://bioinfo.genotoul.fr/multalin). The color code is defined by the MultAlin program. Predicted Fe(II)- and 2OG-binding sites are highlighted.
Thymine 7-hydroxylase (THase), also a member of 2OG-dependent dioxygenases, acts as a key enzyme in the thymidine salvage pathway in fungi (e.g., Neurospora crassa) and catalyzes three sequential oxidation reactions to convert thymine (T) to iso-orotate, which is subsequently processed by a decarboxylase to produce uracil (U) (Smiley et al. 2005; Neidigh et al. 2009). Based on the similar chemistry between 5mC oxidation and the T-to-U conversion, we previously hypothesized that, in theory, Tet proteins should be able to further oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (Wu and Zhang 2010). However, previous studies failed to detect these predicted enzymatic products, likely due to limitations of the assay employed (Tahiliani et al. 2009; Ito et al. 2010). Indeed, with modified experimental conditions, our group and others have recently shown that Tet proteins are capable of further oxidizing 5hmC to 5fC and 5caC (He et al. 2011; Ito et al. 2011). Importantly, 5fC and 5caC can be detected in genomic DNA of mouse ES cells (He et al. 2011; Ito et al. 2011; Pfaffeneder et al. 2011). Consistent with its in vitro activity, overexpression of Tet2 protein in human HEK293 cells, where endogenous Tet protein levels are low, can produce readily detectable levels of 5fC and 5caC in an enzymatic activity-dependent manner (Ito et al. 2011). Conversely, levels of 5fC and 5caC in mouse ES cells are significantly reduced in response to Tet1 knockdown (Ito et al. 2011). These results suggest that Tet protein-mediated oxidation of 5mC may initiate enzymatic cascades that dynamically modify the pre-existing mammalian DNA methylation pattern.
Quantifying and mapping 5mC oxidation derivatives
Cytosine methylation has long been recognized as “the fifth base” in mammalian DNA. Early work noted that the hydroxylated form of 5mC, 5hmC, is present in T-even bacteriophage DNA and is often further modified by glycosylation to protect the phage genome from being degraded by restriction enzymes present in infected hosts (Wyatt and Cohen 1952; Vrielink et al. 1994). The recent discoveries of the sixth base, 5hmC, by the Heintz and Rao groups (Kriaucionis and Heintz 2009; Tahiliani et al. 2009) and of additional 5mC oxidation derivatives (5fC and 5caC) by our and other groups (He et al. 2011; Ito et al. 2011; Pfaffeneder et al. 2011) in the mammalian genome have fueled a strong interest in quantifying global levels as well as mapping genomic distribution of these modified cytosine bases in various cell types and tissues.
However, commonly used approaches for DNA methylation studies, including bisulphite genomic sequencing and methylation-sensitive restriction enzyme digestion, cannot discriminate 5hmC from 5mC (Huang et al. 2010; Jin et al. 2010). There is also evidence suggesting that 5caC is interpreted as C after bisulphite treatment and PCR amplification (He et al. 2011). Thus, bisulphite sequencing, the standard technique used in analyzing DNA methylation patterns, cannot distinguish 5mC from its oxidation derivatives. To circumvent such technical challenges, several strategies exploiting specific biochemical or biophysical properties of modified cytosine bases have been developed. These strategies include (1) thin-layer chromatography (TLC) analysis of modified nucleotides (Ito et al. 2010; Kriaucionis and Heintz 2009; Tahiliani et al. 2009), (2) mass spectrometry (MS) analysis (Ito et al. 2011; Munzel et al. 2010; Globisch et al. 2011), (3) modification-specific antibodies (Ficz et al. 2011; Williams et al. 2011; Wu et al. 2011a), (4) selective chemical labeling of modified cytosine (Szwagierczak et al. 2010; Song et al. 2011; Pastor et al. 2011), and (5) real-time sequencing (Fig. 2; Flusberg et al. 2010).

Detection methods of 5mC oxidation derivatives. (A) Schematic diagrams of in vitro biochemical assays followed by 2D TLC. The spots corresponding to 5mC and its oxidation products are indicated in a representative 2D TLC plate. (B) Flow chart of the procedures used for quantifying 5mC oxidation derivatives in mouse genomic DNA. Representative data of high-performance LC (HPLC) and LC-MS/MS assays are shown. (C) Schematic representations of how antibodies specific to 5mC oxidation products can be used in immunoprecipitation or immunostaining assays. (D) Schematic representation of the 5hmC glucosylation reaction catalyzed by β-glucosyltransferase (β-GT) of T-even bacteriophages.
TLC assay
TLC is a classic method that separates different nucleotides or different modified forms of the same nucleotide based on their differential migration rates on TLC plates. Using the methylation-insensitive restriction enzyme MspI and a TLC assay, Tahiliani et al. (2009) estimated that the 5hmC level in mouse ES cells is ∼0.03% of total nucleotides. Upon leukemia inhibitory factor (LIF) withdrawal, 5hmC levels are reduced to ∼40% in differentiated mouse ES cells, consistent with a concomitant decrease in Tet1 and Tet2 mRNA levels (Koh et al. 2011). Using a two-dimensional (2D) TLC method and nearest-neighbor analysis, Kriaucionis and Heintz (2009) reported that 5hmC level is ∼0.6% and 0.2% of total nucleotides in Purkinje and granule neurons, respectively. However, under standard TLC buffer conditions, 5hmC and 5fC have almost identical migration patterns, and 5caC fails to migrate (Ito et al. 2011). To overcome this technical issue, we developed a modified 2D TLC assay in conjunction with the use of TaqI, a restriction enzyme that is insensitive to all cytosine modifications, allowing for clear separation of 5mC and its oxidation products (Fig. 2A; Ito et al. 2011).
Liquid chromatography (LC) and MS
To better quantify the global levels of 5mC oxidation derivatives, several groups have developed MS-based methods. Using stable isotope-labeled reference compounds and LC-MS, Gobisch et al. (2011) found that 5mC levels are relatively similar in all tested mouse tissues (∼4.3% of all cytosine or 43 × 103 5mC in every 106 C). However, 5hmC levels varied significantly between different tissues, with the highest levels of 5hmC found in various brain tissues (3 × 103 to 7 × 103 5hmC in every 106 C); medium levels in kidney, nasal epithelia, bladder, heart, muscle, and lung (1.5 × 103 to 1.7 × 103 5hmC in every 106 C); and low levels in pituitary gland, liver, spleen, and testes (0.3 × 103 to 0.6 × 103 5hmC in every 106 C) (Munzel et al. 2010; Globisch et al. 2011). Using a similar approach, the same group recently discovered that 5fC is also present in mouse ES cells and estimated that 5%–10% of 5hmC in mouse ES cell DNA is further oxidized to 5fC (0.2 × 103 5fC in every 106 C vs. 3.9 × 103 5hmC in every 106 C) (Pfaffeneder et al. 2011). Upon differentiation by LIF withdrawal or in Dnmt3a/3b-deficient mouse ES cells, 5fC level is significantly reduced, reinforcing the notion that 5fC is generated from pre-existing 5mC/5hmC by Tet proteins. Using a quantitative LC-MS assay, our group recently characterized the enzymatic kinetics of the Tet-catalyzed iterative oxidation reaction and quantified the endogenous levels of 5hmC, 5fC, and 5caC (Fig. 2B; Table 1). This analysis indicated that the genomic content of these modified bases in mouse ES cells is ∼1.3 × 103 5hmC, 20 5fC, and three 5caC in every 106 C, respectively (Ito et al. 2011). While 5hmC and 5fC are also present in various mouse tissues, 5caC can only be reliably detected in mouse ES cells (Table 1; Ito et al. 2011).
Quantification of 5mC and its oxidation derivatives in genomic DNA of HEK293 cells, and mouse ES cells and tissues
Modification-specific antibodies
DNA methylation patterns can be determined by using antibodies against 5mC or methyl-CpG-binding domains (MBDs). Similarly, antibodies specific for 5hmC, which are available from both commercial (e.g., Diagenode and Active Motif) and academic (Williams et al. 2011) sources, have been used for genome-wide mapping of 5hmC distribution by high-throughput sequencing or whole-genome tiling microarrays (Fig. 2C; Ficz et al. 2011; Jin et al. 2011; Stroud et al. 2011; Szulwach et al. 2011; Wu et al. 2011a; Y Xu et al. 2011). Antibodies specific for 5mC and 5hmC are also extensively used in immunostaining assays to examine cellular levels of these epigenetic marks in various biological systems, such as zygotes, developing embryos, and adult brains (Ficz et al. 2011; Iqbal et al. 2011; Ruzov et al. 2011; Wossidlo et al. 2011). In addition, Pastor et al. (2011) took advantage of the fact that bisulphite treatment converts 5hmC into cytosine 5-methylenesulphonate (CMS) and developed specific antisera against CMS. As specific antibodies against 5fC or 5caC become available, they can be used in a similar way to determine the cellular levels as well as the genome-wide location of these newly identified 5mC oxidation derivatives.
Chemical labeling of 5mC oxidation derivatives
While the antibody-based detection methods for 5mC oxidation derivatives are convenient to use and advantageous in immunostaining assays, it has been reported that the immunoprecipitation efficiency of 5hmC antibodies are partially dependent on the density of CpG sites (Pastor et al. 2011). Therefore, sparsely distributed 5hmC marks in genomic DNA may not be readily detected using the 5hmC antibody. To further improve the sensitivity of 5hmC detection methods, several groups developed selective chemical labeling approaches for 5hmC by exploiting the naturally occurring glucosylation process of 5hmC by β-glucosyltransferase (β-GT) in T-even bacteriophages (Fig. 2D; Szwagierczak et al. 2010; Pastor et al. 2011; Song et al. 2011). Such strategy allows for the addition of a glucose moiety to the hydroxyl group of 5hmC using β-GT to yield β-glucosyl-5-hmC. For instance, Song et al. (2011) used purified β-GT to transfer a custom-synthesized UDP-glucose analog (UDP-6-N3-glucose) onto 5hmC. With an azide group present in the chemically modified glucose, a biotin group or other tags can be attached to 5hmC using click chemistry for various applications (Song et al. 2011). A similar approach, termed GLIB (glucosylation, periodate oxidation, and biotinylation), was developed by Rao and colleagues (Pastor et al. 2011). In vitro binding assays suggest that these chemical labeling methods may possess enhanced sensitivity for detecting 5hmC marks sparsely distributed in genomic DNA (Pastor et al. 2011). Importantly, chemical labeling strategy can also be used to add biotin tags to 5fC and 5caC (Ito et al. 2011; Pfaffeneder et al. 2011), providing a potential method to compare the relative abundances of different 5mC oxidation derivatives.
Single-molecule real-time (SMRT) sequencing
While modification-specific antibody or chemical labeling methods can detect the presence of 5mC oxidation derivatives within genomic DNA fragments (200- to 500-base-pair [bp] resolution), neither of the above approaches provides the modification status of cytosine at single-base-pair resolution. Thus, it is challenging to precisely map 5mC and its derivatives when these modified bases are within the same DNA fragments. A recent study has reported promising results on the development of SMRT sequencing, which uses the capacity of DNA polymerase to incorporate C, 5mC, and 5hmC with different kinetics (Flusberg et al. 2010). While the robustness of this technology as well as whether this technology can discriminate 5hmC from 5fC and 5caC remain to be seen, real-time sequencing technologies provide a potential tool to map the various cytosine derivatives at base-pair resolution without the need for bisulphite treatment.
Tet-mediated 5mC oxidation and DNA demethylation
Although the biological significance of Tet-mediated oxidation of 5mC is largely unclear, the relative abundance of 5mC oxidation derivatives such as 5hmC in genomic DNA suggests that these modified cytosine bases may play important roles in modulating 5mC-dependent gene regulatory and biological functions. Indeed, MBD-containing proteins such as methyl-CpG-binding protein 2 (MeCP2) do not recognize 5hmC (Valinluck et al. 2004; Jin et al. 2010), thereby providing a potential means to reverse the gene silencing effect mediated by promoter DNA methylation-dependent recruitment of MBD proteins and their associated corepressor protein complexes (Nan et al. 1998; Feng and Zhang 2001). In addition, 5mC oxidation derivatives such as 5hmC may regulate local chromatin structure by directly recruiting specific DNA-binding proteins. Interestingly, in vitro DNA-binding and molecular simulation analyses suggest that the SET- and RING-associated (SRA) domain of Uhrf1 protein, a key factor in methylation maintenance, binds to not only hemimethylated but also hemi- or fully hydroxymethylated CpG sites (Frauer et al. 2011), suggesting a role for Uhrf1 in mediating 5hmC-dependent functions.
Emerging evidence suggests that Tet protein-mediated 5mC oxidation may also contribute to dynamic changes in global or locus-specific 5mC levels by facilitating both passive and active DNA demethylation. Because 5hmC is not recognized by Dnmt1 during DNA replication (Valinluck and Sowers 2007), conversion of 5mC to 5hmC will prohibit the maintenance of existing DNA methylation patterns and lead to passive DNA demethylation in proliferating cells. This replication-dependent dilution mechanism of 5hmC may be important for genome-wide erasure of 5mC in the paternal genome in zygotes and preimplantation embryos (see below). Furthermore, 5mC oxidation derivatives may represent intermediate products in the process of replication-independent active DNA demethylation (enzymatic conversion of 5mC to C). Although it is well established that plants (e.g., Arabidopsis thaliana) use the Demeter (DME)/repressor of silencing 1 (ROS1) family of 5mC glycosylases and the base excision repair (BER) pathway to achieve active DNA demethylation (Zhu 2009), a mammalian ortholog of the DME/ROS1 family of glycosylases has yet to be identified. How active DNA demethylation takes place in mammals has been controversial (Ooi and Bestor 2008; Wu and Zhang 2010). The discovery of 5mC oxidation derivatives in mammalian cells immediately raises the possibility that Tet proteins may play a role in the process of active DNA demethylation (Tahiliani et al. 2009; Wu and Zhang 2010). Here we discuss existing biochemical, genetic, and functional evidences for three proposed enzymatic pathways of active DNA demethylation initiated by Tet proteins (Fig. 3).
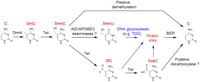
Proposed models of Tet-initiated DNA demethylation pathways. DNA methylation (5mC) is established and maintained by DNA methyltransferases (Dnmt). In mammals, 5mC can be hydroxylated by the Tet family of dioxygenases to generate 5hmC. 5hmC is recognized poorly by Dnmt1 and can lead to replication-dependent passive demethylation. 5hmC can be further oxidized by Tet proteins to produce 5fC and 5caC. Alternatively, 5hmC may be further deaminated to become 5hmU by AID/APOBECC deaminases. 5hmU, 5fC, and 5caC can be excised from DNA by glycosylases such as TDG. A putative decarboxylase may directly convert 5caC to C. Cytosine and its derivatives are highlighted in red. DNA glycosylase TDG-catalyzed reactions are indicated by blue arrows.
Iterative 5mC oxidation followed by DNA glycosylase/BER
As discussed above, Tet proteins can generate 5hmC, 5fC, and 5caC through iterative oxidation of 5mC (He et al. 2011; Ito et al. 2011). These 5mC oxidation derivatives can potentially serve as substrates for mammalian DNA glycosylases or deaminases (see below). Indeed, recent studies have demonstrated that thymine DNA glycosylase (TDG) can efficiently excise 5fC or 5caC in the context of CpG sites (He et al. 2011; Maiti and Drohat 2011). Thus, subsequent repair of the resulting abasic site would regenerate unmethylated cytosine. Interestingly, TDG removes 5fC from the G:5fC substrate at a rate even faster (∼40%) than processing the G:T mismatch (Maiti and Drohat 2011), probably due to the weakened N-glycosidic bond between the nucleobase and the sugar (Bennett et al. 2006). In contrast, TDG has essentially no activity for 5hmC (Maiti and Drohat 2011). The robust excision activity of TDG toward 5fC and 5caC may be one explanation as to why 5fC and 5caC are present at very low levels in the genome compared with 5hmC (Ito et al. 2011; Pfaffeneder et al. 2011). In agreement with the proposal that TDG promotes active demethylation, deletion of TDG in mice leads to embryonic lethality and aberrantly elevated DNA methylation levels at a cohort of gene promoters (Cortazar et al. 2011; Cortellino et al. 2011). In contrast, other DNA glycosylases, such as single-strand-selective monofunctional U DNA glycosylase 1 (SMUG1) and MBD protein 4 (MBD4), have no significant activity for excision of 5fC and 5caC from DNA (He et al. 2011; Maiti and Drohat 2011), suggesting that TDG might be the only mammalian DNA glycosylase to have robust excision activity for 5fC and 5caC. Thus, these findings lead to a multistep active DNA demethylation model linking Tet1-mediated iterative oxidation of 5mC and the BER pathway initiated by the TDG DNA glycosylase (Fig. 3). However, it is important to note that although depletion of TDG in mouse ES cells resulted in an increase in the 5caC level (He et al. 2011), the amount of estimated 5caC under TDG-depleted conditions is still below the level of 5fC under normal conditions (He et al. 2011; Ito et al. 2011). Given the high efficiency of full-length Tet2 in converting 5mC to 5caC (He et al. 2011), it is anticipated that 5caC would accumulate at a much higher level under TDG depletion conditions if TDG is the only enzyme capable of processing 5caC. Therefore, it is possible that additional enzymes that possess 5caC processing activity exist in mammalian cells.
5hmC deamination followed by DNA glycosylase/BER
Another potential active DNA demethylation pathway that can be initiated by Tet proteins involves deamination of 5hmC followed by DNA repair. In this scenario, 5hmC may first be deaminated by the AID (activation-induced deaminase)/APOBEC (apolipoprotein B mRNA-editing enzyme complex) family of cytidine deaminases to produce 5-hydroxymethyluracil (5hmU), followed by 5hmU:G mismatch repair through the action of DNA glycosylases and the BER pathway (Fig. 3). This hypothesis is supported by recent studies demonstrating that DNA glycosylases such as TDG and SMUG1 exhibit robust excision activity against 5hmU:G in dsDNA (Cortellino et al. 2011), whereas they have no or very low 5hmC glycosylase activity in an in vitro biochemical assay (Cortellino et al. 2011; He et al. 2011). Furthermore, studies in human HEK293 cells demonstrated that TDG can directly interact with AID (Cortellino et al. 2011), suggesting that the deamination and DNA glycosylase activity may be coupled.
However, one open question that remains to be addressed is to what extent the AID/APOBEC family of cytidine deaminases is used to deaminate 5hmC to generate 5hmU in vitro and in vivo (Fig. 3). Whereas AID and APOBEC1 are shown to possess the capacity to deaminate 5mC to T in the context of ssDNA in vitro (Morgan et al. 2004), no direct biochemical evidence suggests that these deaminases exhibit robust activity for 5hmC. In addition, mass spectrometric analysis indicates that 5hmU, the predicted intermediate of this pathway, does not accumulate to a detectable level in mammalian cells (Globisch et al. 2011; Pfaffeneder et al. 2011). This indicates that either the deamination reaction does not occur extensively in vivo or 5hmU is extremely short-lived. In agreement with this, although AID may contribute to active PGC demethylation in vivo (Popp et al. 2010), it is probably responsible for only a small portion of this large-scale demethylation process, as substantial demethylation still occurs in the absence of AID. Nevertheless, 5hmC deamination followed by the BER pathway does seem to be a potential mechanism underlying loci-specific active demethylation. For example, a recent study has suggested that Tet1 and AID may cooperate in demethylating a methylated DNA duplex (Guo et al. 2011). In this study, cotransfection of plasmid DNA encoding various AID/APOBEC deaminases with a linearized 5mC- or 5hmC-containing reporter into HEK293 cells resulted in locus-specific demethylation of 5hmC- but not 5mC-containing dsDNA (Guo et al. 2011). Interestingly, Guo et al. (2011) also observed that 5hmC in a non-CpG context is more frequently demethylated than those in CpG dinucleotides (∼9% for 5hmCpHs vs. ∼2% for 5hmCpGs), and 5hmC in the nontranscribed strand seems to be preferentially demethylated.
Iterative 5mC oxidation followed by decarboxylation
Both mechanisms described above involve glycosylases and the DNA repair pathway. Such mechanisms are unlikely to be used in large-scale DNA demethylation processes observed in PGCs and zygotes, as genome-wide repair of mismatches in such a limited time window will put tremendous pressure on genome stability. Therefore, we previously proposed a simple mechanism involving Tet-mediated iterative oxidation followed by decarboxylation (Wu and Zhang 2010). Given that only two enzymes are required to convert 5mC to C in this hypothetical mechanism (Fig. 3), it obviates the need for a DNA strand break that is required for DNA repair-based mechanisms, thereby relieving the pressure of genome instability associated with BER. Despite the simplicity of this proposed demethylation pathway and the precedent of a decarboxylase in thymidine salvage reactions, the existence of the putative decarboxylase capable of removing carboxyl group from 5caC to regenerate unmodified cytosine remains to be revealed.
Genomic distribution of Tet1 and 5hmC, and their role in transcriptional regulation
A prevailing view of the role of DNA methylation in gene regulation is that DNA methylation primarily functions in long-term gene silencing. Indeed, DNA methylation at the gene promoter can either block the binding of transcriptional factors or facilitate the recruitment of MBD-associated corepressor complexes (Bird 2002). However, recent genome-wide methylome analyses in pluripotent stem cells and differentiated cells indicate that not only is promoter methylation dynamic during cellular differentiation, but actively transcribed genes tend to be associated with high levels of gene body DNA methylation (Meissner et al. 2008; Suzuki and Bird 2008; Lister et al. 2009). A recent genome-wide mapping and functional study of the de novo DNA methyltransferase Dnmt3a also provide evidence supporting the view that DNA methylation may play a role in both gene repression and activation (Wu et al. 2010). These genome-wide DNA methylation studies have therefore revealed a more complex, context-dependent role for DNA methylation in transcriptional regulation.
To better understand the gene regulatory function of Tet proteins and 5mC oxidation, we and others recently mapped distribution of Tet1 and 5hmC across the genome of mouse and human ES cells (Ficz et al. 2011; Pastor et al. 2011; Stroud et al. 2011; Szulwach et al. 2011; Williams et al. 2011; Wu et al. 2011a,b; Y Xu et al. 2011). Comparative analysis of data sets from different laboratories indicates that different Tet1 antibodies and 5hmC mapping methods generated similar results (Wu and Zhang 2011). Using pluripotent ES cells as a model system, these genome-wide analyses uncovered several key features of genomic occupancy of Tet1 and 5hmC as well as their potential functions in transcriptional regulation (Fig. 4).
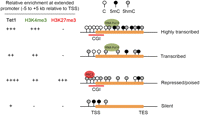
Relationship of Tet1/5hmC and transcription in mouse ES cells. Schematic representation of the relative enrichment of Tet1 and major histone modifications (H3K4me3 and H3K27me3), as well as the distribution of 5mC/5hmC at four major classes of genes: (1) Highly transcribed genes with a CpG-rich promoter are associated with high levels of Tet1 and H3K4me3 at their promoters, as well as high levels of 5mC/5hmC within their gene bodies (particularly at 3′ intragenic regions). (2) Actively transcribed genes with CpG-poor promoters (e.g., a subset of pluripotency factors such as Nanog and Tcl1) are associated with medium/low levels of Tet1 and 5hmC at their promoters as well as within their gene bodies. (3) Polycomb-repressed yet “poised” genes (e.g., lineage-specific transcription factors) are enriched with high levels of Tet1, 5hmC, H3K4me3, and H3K27me3 at their extended promoters. (4) Silent genes with a CpG-poor promoter (e.g., tissue-specific genes) are generally associated with sparsely distributed 5mC and 5hmC at their proximal promoter, but are devoid of high levels of Tet1 and PRC2. (TSS) Transcriptional start site; (TES) transcriptional end site.
First, Tet1 exhibits a strong preference for genomic regions with densely clustered CpG dinucleotides, termed as CpG islands (CGIs) (Williams et al. 2011; Wu et al. 2011b; Y Xu et al. 2011), which are generally free of DNA methylation and frequently overlapped with transcriptional start sites (Deaton and Bird 2011). This is possibly due to the presence of a CpG-binding CXXC zinc finger domain in Tet1 (Fig. 1; Iyer et al. 2009; Zhang et al. 2010). Interestingly, the CXXC domain has also been found in other epigenetic regulators such as Cfp1 (CXXC finger protein 1) and the H3K36me2 (dimethylated H3K36) histone demethylase Kdm2a that are enriched at unmethylated CGIs (Blackledge et al. 2010; Thomson et al. 2010), raising the possibility that these CXXC domain-containing proteins may cooperate in contributing to the establishment of a transcriptionally permissive chromatin state at CGIs (Deaton and Bird 2011). An attractive scenario is that, at a majority of CGIs, Cfp1 promotes trimethylation of Lys 4 on histone H3 (H3K4me3) via recruitment of the H3K4me3 methyltransferase Setd1 (Thomson et al. 2010), Kdm2a reduces the level of H3K36me2 (Blackledge et al. 2010), and Tet1 maintains a DNA hypomethylated state by actively removing any sporadic de novo DNA methylation. In support of this model, Tet1-bound CGIs are DNA hypomethylated, and Tet1 deficiency results in an increase in 5mC levels at many Tet1-enriched regions (Wu et al. 2011b; Y Xu et al. 2011).
Second, both Tet1 and 5hmC are highly enriched at gene promoters that are associated with bivalent domains (Pastor et al. 2011; Williams et al. 2011; Wu et al. 2011a,b; Y Xu et al. 2011), which are marked with the transcriptional permissive mark H3K4me3 as well as the Polycomb repression complex 2 (PRC2) deposited repressive mark H3K27me3. Bivalent gene promoters are generally associated with poised developmentally regulated genes, particularly lineage-specific transcription factors, suggesting a role for Tet1 and 5hmC in promoting a repressive but “poised” state at these gene promoters. In agreement with this model, depletion of Tet1 in mouse ES cells results in impaired recruitment of Ezh2 (a core subunit of PRC2) to bivalent gene promoters (Wu et al. 2011b), indicating that Tet1 and 5hmC may functionally contribute to the maintenance of the undifferentiated state of mouse ES cells by facilitating PRC2-mediated repression of lineage-specific genes. Interestingly, although 5mC at promoters is also linked to gene repression, 5mC is not enriched at bivalent gene promoters (Fouse et al. 2008). Thus, 5hmC and 5mC may have different regulatory functions at gene promoters.
Third, Tet1 and 5hmC seem to play dual functions in transcription regulation. Chromatin immunoprecipitation (ChIP) combined with sequencing (ChIP-seq) analyses from multiple laboratories have revealed that Tet1 binds to promoters of highly transcribed as well as PRC2-repressed genes (Wu and Zhang 2011). Genome-wide expression profiling of control and Tet1-depleted mouse ES cells also indicates that Tet1 may have both repressive and activating functions on its direct target genes (Williams et al. 2011; Wu et al. 2011b; Y Xu et al. 2011). A significant fraction of genes aberrantly up-regulated in Tet1-depleted mouse ES cells are Tet1/PRC2-cobound and transcriptionally repressed lineage-specific genes (Wu et al. 2011b), whereas a cohort of actively transcribed Tet1 target genes, including several pluripotency-related transcription factors (e.g., Nanog, Tcl1, and Esrrb), are found to be down-regulated in the absence of Tet1 (Ficz et al. 2011; Wu et al. 2011b). In addition, 5hmC has been shown to be enriched in both the gene body of highly transcribed genes (particularly at exons) and the promoters of PRC2-repressed genes (Ficz et al. 2011; Pastor et al. 2011; Williams et al. 2011; Wu et al. 2011a; Y Xu et al. 2011), further supporting a potential role for 5hmC in both transcriptional repression and activation (Robertson et al. 2011). Interestingly, Williams et al. (2011) also showed that Tet1 associates with the Sin3A corepressor complex to repress a subset of Tet1 targets, and the transcriptional repression function of Tet1 appears to be independent of its enzymatic activity in mouse ES cells. Further studies are needed to determine whether the enzymatic activity-independent transcriptional function of Tet1 is specific to pluripotent stem cells or is a general mechanism that also operates in other cell types.
Finally, 5hmC, but not 5mC, is enriched at many intergenic cis-regulatory elements, such as active enhancers and insulator-binding sites (Ficz et al. 2011; Pastor et al. 2011; Williams et al. 2011; Wu et al. 2011a). Recent genome-wide mapping studies of 5hmC in human ES cells also confirmed that 5hmC is enriched at binding sites of many transcriptional factors related to pluripotency (Stroud et al. 2011; Szulwach et al. 2011), indicating a conserved role for Tet1 and 5hmC in controlling gene expression through regulation of enhancer functions.
Together, genome-wide studies in undifferentiated ES cells and during ES cell differentiation have provided detailed information regarding the distribution of Tet1 and 5hmC and supported the notion that Tet1-mediated gene regulation plays an important role in orchestrating the balance between the pluripotent state and initiation of cellular differentiation. Genome-wide analysis of other 5mC oxidation derivatives is likely to provide additional insights into the role of Tet proteins in gene regulation and active DNA demethylation.
Biological functions of Tet proteins in development and diseases
Although all Tet family members (Tet1–3) possess 5mC oxidation activity, their expression levels in various cell types and tissues are very different (Ito et al. 2010). For example, Tet1 and Tet2 are highly expressed in mouse ES cells, but Tet3 is more enriched in oocytes and one-cell zygotes. The distinct expression pattern of Tet1–3 suggests that these proteins may play nonoverlapping biological functions in a developmentally regulated and tissue-specific manner.
Role of Tet3-mediated 5mC oxidation in zygotic DNA demethylation
While DNA methylation patterns are relatively stable in somatic cells, rapid erasure of global DNA methylation patterns has been observed in developing PGCs as well as zygotes (Sasaki and Matsui 2008; S Feng et al. 2010; Wu and Zhang 2010). In one-cell zygotes, the 5mC mark in the paternal pronucleus quickly disappears before the first cell division (depicted in orange in the top panel of Fig. 5; Mayer et al. 2000; Oswald et al. 2000), whereas the maternal genome is proposed to be protected from zygotic demethylation by DNA-binding proteins, such as Stella/Dppa3 (Nakamura et al. 2007). Using specific antibodies against 5hmC or 5mC, recent studies have shown that the paternal and maternal genomes of mouse zygotes at late pronuclear (PN4–5) stages are predominantly marked by 5hmC and 5mC, respectively, and that appearance of 5hmC (around PN3 stage) coincides with the loss of 5mC marks in the paternal genome (summarized in the bottom panel of Fig. 5; Iqbal et al. 2011; Wossidlo et al. 2011). The asymmetric marking of 5hmC in the paternal pronucleus is also observed in bovine and rabbit zygotes (Wossidlo et al. 2011), suggesting a conserved role for oxidation of 5mC in the erasure of paternal 5mC immediately after fertilization. Gene expression analysis indicated that Tet3, but not Tet1 and Tet2, is expressed at high levels in oocytes and zygotes, but rapidly decreases at two-cell and later stages (Iqbal et al. 2011; Wossidlo et al. 2011). In agreement with the notion that Tet3 is responsible for 5mC oxidation in the paternal pronucleus, either conditional deletion of Tet3 from the female germ cells or siRNA-mediated down-regulation of zygotic Tet3 impedes the conversion of 5mC to 5hmC in the paternal genome (Gu et al. 2011; Wossidlo et al. 2011). Furthermore, Tet3 protein appears to be specifically enriched in the paternal pronucleus at the zygotic stage (Gu et al. 2011). Subsequent bisulphite sequencing analysis suggests that zygotic Tet3 may be required for active DNA demethylation at specific loci in the paternal genome, including Line1 transposons and gene regulatory regions of pluripotency factors (Oct4 and Nanog), which are hypermethylated in sperm. However, loss of Tet3 does not affect the methylation status of several imprinting loci (Gu et al. 2011). Interestingly, Tet3 is dispensable for germ cell maturation, fertilization, and preimplantation development, but deletion of Tet3 from either PGCs (TNAP-Cre) or growing oocytes (Zp3-Cre) resulted in increased frequency of developmental failure of embryos, possibly due to delayed/attenuated activation of the paternal allele of genes important for embryogenesis (Gu et al. 2011). In addition, somatic cell nuclear transfer (SCNT) using Tet3-deficient oocytes, but not control oocytes, resulted in failure in 5mC oxidation of somatic genomes, impaired demethylation at the somatic Oct4 promoter, and activation of an Oct4-EGFP transgene (Gu et al. 2011). Together, these results suggest that Tet3 acts as a critical factor for zygotic epigenetic reprogramming by initiating the global conversion of 5mC to 5hmC and possibly other oxidation forms.
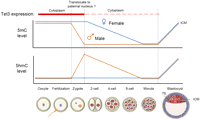
Dynamic changes of 5mC and 5hmC levels in the paternal and maternal genomes during preimplantation development. Tet3 is highly expressed in the oocyte and one-cell zygote. Immediately after fertilization, Tet3 may potentially relocate from the cytoplasm to the paternal nucleus to convert 5mC to 5hmC. Subsequently, paternal and maternal genomes undergo replication-dependent dilution of 5hmC and 5mC, respectively. It is important to note that replication-independent active DNA demethylation may occur at specific loci, but the exact mechanism is currently unclear. New DNA methylation patterns in the ICM are re-established by de novo DNA methyltransferases Dnmt3a and Dnmt3b. (TE) Trophectoderm.
Interestingly, asymmetric marking of 5hmC in sperm-derived chromosomes persists into two-cell stage embryos (Iqbal et al. 2011), suggesting that 5hmC is not rapidly removed on a genome-wide scale. Thus, it is important to determine the fate of paternal 5hmC during preimplantation development. As discussed above (Fig. 3), 5hmC/5fC/5caC can be actively processed through enzymatic reactions or be passively diluted through DNA replication. In a recent study, careful analysis of high-resolution mitotic chromosome spreads that are positive for 5hmC staining at the one-, two-, four-, and eight-cell stages indicates that 5hmC marks on the paternal chromosomes are gradually lost from the one- to eight-cell stage, and this process appears to be mediated by replication-dependent dilution, as, on average, half of the chromosomes retain the 5hmC mark after each round of cell division (Inoue and Zhang 2011). The above results support the model in which global erasure of paternal 5mC is first initiated by Tet3-mediated conversion of 5mC to 5hmC in the male pronucleus, followed by replication-dependent passive loss of 5hmC during preimplantation development. Similarly, a replication-dependent passive demethylation mechanism has been proposed to be responsible for global loss of 5mC marks on the maternal chromosomes (Rougier et al. 1998). While these findings cannot exclude the possibility that 5mC oxidation derivatives may also serve as intermediates for replication-independent active DNA demethylation pathways (Fig. 3), whether and how specific loci in paternal and maternal genomes undergo actual DNA demethylation (conversion of 5mC/5hmC to C, not just to 5fC or 5caC) in zygotes and preimplantation embryos awaits further investigation, as bisulphite sequencing analysis cannot discriminate 5caC from unmethylated cytosine (He et al. 2011). Of note, a recent study identified the elongator complex as a potential regulator of zygotic paternal genome demethylation (Okada et al. 2010; Wu and Zhang 2010). It will be of interest to determine whether elongator and Tet3 proteins cooperate to mediate the erasure of paternal 5mC. Given that developing PGCs also undergo global erasure of 5mC (Hajkova et al. 2002; Sasaki and Matsui 2008), it will be interesting to determine whether Tet-mediated 5mC oxidation is also involved in PGC demethylation.
Role of Tet1 in regulating pluripotency and lineage differentiation of mouse ES cells
Concomitant with the rapid reduction of Tet3 expression at the two-cell stage, the expression of Tet1 is rapidly up-regulated at later preimplantation stages (Iqbal et al. 2011; Wossidlo et al. 2011). As the zygote develops into the blastocyst, Tet1 and Tet2 are expressed at high levels in the inner cell mass (ICM). Consistently, Tet1 and Tet2, but not Tet3, are expressed in mouse ES cells. Upon in vitro differentiation of mouse ES cells, Tet1 and Tet2 expression is rapidly decreased, whereas Tet3 expression is up-regulated (Tahiliani et al. 2009; Ito et al. 2010; Koh et al. 2011). Given that both Tet1 and Tet2 are direct downstream targets of a cohort of pluripotency factors (Koh et al. 2011) and that Tet1 binds to gene promoters of some key ES cell transcription factors (Wu and Zhang 2011), Tet proteins are likely to be part of the pluripotency regulatory circuit.
shRNA-mediated Tet1 knockdown in mouse ES cells suggests that Tet1 deficiency leads to a decrease in total 5hmC levels (∼50%) and increased DNA methylation at the Nanog-proximal promoter, which is accompanied by a decrease in Nanog expression and impaired proliferation of ES cells (Ito et al. 2010). Subsequent genome-wide location analyses in mouse ES cells have confirmed that Tet1 and 5hmC are enriched at promoter regions of several pluripotency factors, including Nanog, Tcl1, and Esrrb (Wu and Zhang 2011). RNA-seq analysis of Tet1 and Tet2 double knockdown mouse ES cells also showed that several genes related to pluripotency are down-regulated in the absence of Tet proteins (Ficz et al. 2011). However, other studies using different sets of shRNAs to down-regulate Tet1 and Tet2 suggest that Tet1/2 deficiency does not affect the expression of pluripotency factors and mouse ES cell proliferation (Koh et al. 2011; Williams et al. 2011). These discrepancies between in vitro experiments are possibly due to differences in mouse ES cell background, culturing conditions, and/or off-target effects of shRNAs (Williams et al. 2011; Wu and Zhang 2011). Further analysis of lineage-specific gene expression and teratomas indicate that Tet1 deficiency leads to increased spontaneous differentiation toward trophoectoderm and mesoendoderm lineages (Ito et al. 2010; Ficz et al. 2011; Koh et al. 2011). The above results, together with recent findings on the dual functions of Tet1 in both Polycomb repression of developmental genes and transcriptional activation of pluripotency genes, suggest that Tet1 is potentially required for orchestrating the balance between pluripotency maintenance and lineage commitment.
To further study the function of Tet1 in ES cell maintenance and in vivo development, Jaenisch and colleagues (Dawlaty et al. 2011) recently generated Tet1-null mouse ES cells and mice. In Tet1-null mouse ES cells, a decrease in 5hmC levels (∼35%) is observed and 221 genes are found to be significantly dysregulated. However, Tet1-null ES cells maintain a normal morphology, express proper levels of key pluripotency factors, and support embryonic development in a tetraploid complementation assay (Dawlaty et al. 2011). While Jaenisch and colleagues (Dawlaty et al. 2011) also observed altered expression of lineage-specific marks (Brachyury and Pax6) in differentiating mutant ES cells and skewed differentiation toward trophectoderm in Tet1-null teratomas, Tet1 mutant ES cells fail to exhibit similar phenotypic defects in vivo. These results suggest that in vitro differentiation defects exhibited in mutant ES cells and teratoma assays are less pronounced in the context of embryonic development in vivo. Further analysis of Tet1-null mice indicate that mutant mice are viable, but 75% of the homozygous mutant pups display a smaller body size at birth, suggesting loss of Tet1 leads to a developmental delay. Moreover, both male and female germ cells are generated in homozygous mutant embryos, but the number of progenies derived from the intercross of Tet1-null males and females are significantly reduced, indicating that either a subset of Tet1-null mice are embryonic lethal or germ cell development is impaired in homozygous Tet1-null mice (Dawlaty et al. 2011). Given that Tet1 is highly expressed in developing PGCs (Hajkova et al. 2010), further investigations are required to elucidate the role of Tet1 in epigenetic reprogramming of PGCs and gametogenesis.
Role of Tet proteins in brain development and activity-dependent DNA demethylation
Brain-specific deletion of DNA methyltransferases results in premature death, neural developmental defects, and impaired neuronal functions (Fan et al. 2001, 2005; Nguyen et al. 2007; J Feng et al. 2010; LaPlant et al. 2010; Wu et al. 2010). Loss-of-function mutations of the X-chromosome-linked gene encoding MECP2 causes Rett syndrome, an autism spectrum disorder predominantly found in girls (Amir et al. 1999). These findings suggest that precise control of DNA methylation patterns is required for proper brain development. Among various mouse cell types and tissues, neurons in the CNS appear to contain the highest levels of 5hmC, almost 10 times higher than that of mouse ES cells, suggesting that Tet proteins may play a critical role in brain development and maturation.
Using a chemical labeling strategy to determine the total amount of 5hmC in the mouse cerebellum at different stages of development, Song et al. (2011) report a gradual increase from postnatal day 7 (P7; 0.1% of all nucleotides) to adult stage (0.4%). They also determined the genome-wide distribution of 5hmC in P7 and adult cerebellum (Song et al. 2011). This analysis identified 5425 genes acquiring 5hmC during aging. Among them, genes related to age-dependent neurodegenerative disorders, angiogenesis, and hypoxia response are enriched. Previous studies show that active DNA demethylation at specific gene promoters (e.g., Bdnf and Fgf1) may be required for activity-dependent adult neurogenesis (Ma et al. 2009; Wu and Sun 2009). A more recent study indicates that Tet1 may contribute to this process by initiating 5mC hydroxylation followed by AID/APOBEC-mediated deamination and BER (Guo et al. 2011). These results raise the possibility that 5hmC may play a critical role in postnatal neural development, age-related neurodegeneration, and transcriptional regulation of activity-dependent neural plasticity.
Role of Tet2 in hematopoiesis and leukemia
Aberrant DNA methylation pattern is one of the hallmarks of cancer cells (Esteller 2008; Gal-Yam et al. 2008). In general, cancer cells display global hypomethylation and promoter hypermethylation of tumor suppressor genes. Consistently, accumulating evidence suggests that somatic mutations in DNA methyltransferases and 5mC-modifying enzymes, such as TET proteins, are associated with oncogenic transformation. For instance, mutations in the de novo DNA methyltransferase DNMT3A were recently found in a significant fraction of patients with AML (Shah and Licht 2011; Yan et al. 2011).
The first evidence implicating TET proteins in tumorigenesis was the identification of TET1 as a rare fusion partner of MLL in patients with AML (Ono et al. 2002; Lorsbach et al. 2003). The chromosome 4q24 harbors the human TET2 gene and displays recurrent microdeletions and copy-neutral loss of heterozygosity in patients with myeloid malignancies (Viguie et al. 2005). In 2009, two studies identified somatic TET2 mutations in patients with myeloproliferative neoplasms (MPNs) and myelodysplastic syndromes (MDSs) (Delhommeau et al. 2009; Langemeijer et al. 2009). Subsequent investigations of larger cohorts of leukemia patients suggest that TET2 mutations are frequently observed in a diverse spectrum of myeloid malignancies, including MDS (19%–26%), MPN (12%–37%), chronic myelomonocytic leukemia (CMML; 20%–50%), de novo AML (7%–23%), and secondary AML (sAML) (Abdel-Wahab et al. 2009; Delhommeau et al. 2009; Langemeijer et al. 2009; Tefferi et al. 2009a,b; Abdel-Wahab 2011; Pronier et al. 2011). Most recently, recurrent TET2 mutations were also identified in B- and T-cell lymphoma (Quivoron et al. 2011). For detailed discussion of clinical aspects of TET2 mutations in hematopoietic malignancies, please refer to a recent review by Aifantis and colleagues (Cimmino et al. 2011).
To better understand the mechanism by which somatic TET2 mutations contribute to leukemia, several studies examined the effect of disease-associated mutations on TET2 catalytic activity and global epigenetic profiles. Ko et al. (2010) demonstrated that TET2 mutations found in patients with myeloid malignancies impair its enzymatic activity. Consistently, genomic DNA derived from patient bone marrows with TET2 mutations contains significantly lower levels of 5hmC compared with that from healthy controls (Ko et al. 2010). Interestingly, samples from patients with low 5hmC levels also display a hypomethylated state relative to controls at the majority of differentially methylated CpG sites (Ko et al. 2010). However, a recent integrative genetic and methylation analysis of a large, homogeneous cohort of AML patients showed that TET2 mutations are predominantly associated with a DNA hypermethylation state (Figueroa et al. 2010). The discrepancies between these two studies are probably due to differences in disease subtypes (diverse myeloid malignancies vs. de novo AML only) and methylation profiling platforms (Illumina Infinium methylation assay vs. HpaII tiny fragment enrichment by ligation-mediated PCR [HELP] assay). Given that both methylation assays used in previous studies only assess a subset of CpG sites at gene promoter regions, unbiased genome-wide mapping of 5mC and 5hmC distribution is required to better understand the extent of alterations in DNA methylation associated with TET2 mutations.
Interestingly, recent mutational analyses of AML patients have shown that TET2 and the isocitrate dehydrogenase gene IDH1/2 mutations are mutually exclusive (Figueroa et al. 2010). IDH1/2 are NADP-dependent enzymes that catalyze the conversion of isocitrate to 2OG. Recurrent somatic mutations of IDH1/2 are frequently found in glioma and AML patients (Parsons et al. 2008; Mardis et al. 2009; Yan et al. 2009; Gross et al. 2010; Marcucci et al. 2010; Ward et al. 2010). Subsequent studies showed that mutant IDH enzymes predominantly acquire a neomorphic function to produce 2-HG, an oncometabolite that impairs catalytic activity of many Fe(II)- and 2OG-dependent dioxygenases, including the JmjC family of histone demethylases (Klose et al. 2006) and the Tet family of DNA hydroxylases. 2-HG inhibits the enzymatic activity of these enzymes by competing with their cosubstrate, 2OG (Dang et al. 2009; Gross et al. 2010; Ward et al. 2010; W Xu et al. 2011). These studies suggest that IDH1/2 mutations may alter DNA methylation patterns by inhibiting TET2 enzymatic activity. Indeed, methylation profiling of samples from AML patients with IDH1/2 mutations has demonstrated that patient samples exhibited elevated DNA methylation at differentially methylated regions when compared with control samples (Figueroa et al. 2010). Collectively, these results suggest that loss-of-function TET2 mutations and neomorphic IDH1/2 mutations in hematopoietic malignancies may share a common disease mechanism and pathoetiology that lead to alteration in 5hmC and 5mC patterns (Fig. 6). Thus, therapies modulating catalytic activity of TET proteins might be beneficial for the treatment of patients with TET2 mutations.

Leukemic mutations of TET2 or IDH1/2 lead to altered 5mC and 5hmC patterns at tumor suppressor gene promoters. TET2 mutations that impair the conversion of 5mC to 5hmC may cause increased DNA methylation at tumor suppressor gene promoters. Neomorphic mutations of IDH proteins also inhibit TET2 enzymatic activity by producing oncometabolite 2-HG (denoted as a triangle), which competes with 2OG (denoted as a circle) for TET2. Aberrant methylation of gene promoters may result in decreased transcription of tumor suppressors. Notably, TET2 and IDH mutations are mutually exclusive in AML patients.
Frequent mutations of TET2 in leukemia patients suggest that TET2 may function as a physiological regulator of hematopoiesis. To elucidate the functions of TET2 in hematopoietic differentiation and homeostasis, several groups have used various genetic approaches to investigate the effect of Tet2 deficiency on normal hematopoiesis. The approaches include shRNA-based Tet2 knockdown and targeted deletion of Tet2 in mice (Figueroa et al. 2010; Ko et al. 2010, 2011; Li et al. 2011; Moran-Crusio et al. 2011; Quivoron et al. 2011). Despite different phenotypic results between reports using in vitro knockdown of Tet2 (Figueroa et al. 2010; Ko et al. 2010; Moran-Crusio et al. 2011), all Tet2-null mouse models generated highly similar phenotypes. Although all three Tet family proteins are expressed in hematopoietic systems, deletion of Tet2 alone is sufficient to cause significant loss of 5hmC in genomic DNA. Importantly, Tet2 deletion led to gradual enlargement of the stem/progenitor cell pool in a cell-autonomous manner, as shown by increased expression of stem cell marker genes (e.g., c-Kit) as well as markedly elevated self-renewal capacity. Intriguingly, all Tet2-null mouse models progressively develop myeloid neoplasms, despite minor differences in disease onset time and progression kinetics. Consistent with the high frequency of TET2 mutations in human CMML, Tet2-null mouse models exhibited predominantly CMML-like disease. Furthermore, functional analysis showed that Tet2 haploinsufficiency is sufficient to confer increased self-renewal to stem/progenitor cells and promote myeloproliferation in vivo (Moran-Crusio et al. 2011; Quivoron et al. 2011), suggesting clinically relevant gene dosage effects. Thus, Tet2 plays a critical role in regulating normal hematopoietic differentiation and homeostasis, and deletion of Tet2 is sufficient to initiate myeloid transformation.
Concluding remarks and perspectives
Since the discovery of Tet family of proteins in 2009, remarkable progress has been made in characterizing their enzymatic activities, developing methods for detection of newly identified enzymatic products, and understanding the role of Tet-mediated 5mC oxidation in DNA demethylation and transcriptional regulation. Genetic deletion analysis in mice has further revealed that Tet proteins play essential roles in zygotic epigenetic reprogramming, stem cell differentiation, hematopoiesis, and the development of myeloid malignancies.
Despite the significant progress, several key questions about the mechanisms and functions of Tet proteins are yet to be answered. First, current data suggest that Tet-mediated oxidation of 5mC is involved in both passive and active DNA demethylation. While 5mC hydroxylation can clearly lead to replication-dependent global erasure of 5mC in the paternal pronucleus, multiple enzymatic pathways (e.g., 5mC oxidation/DNA glycosylase, AID/APOBEC deaminase-mediated 5hmC deamination/DNA glycosylase, and 5mC oxidation/putative decarboxylase) might be used to complete the process of active DNA demethylation initiated by Tet proteins. Thus, additional investigations are needed to define the physiological context, mechanistic details, and relative importance of each of the potential pathways in the process of active DNA demethylation. Second, genome-wide analysis of Tet1 and 5hmC distribution in mouse ES cells suggests that Tet proteins may have dual functions in transcriptional regulation. It is currently unclear how deposition of 5hmC and/or other oxidation derivatives at different regulatory regions (e.g., promoters, gene body, and enhancers) can contribute to distinct transcriptional states. Third, given that Tet3 may potentially translocate from the oocyte cytoplasm to the paternal pronucleus at the zygotic stage and that 5hmC accumulates to significant levels at specific genomic regions, it is likely that associating factors and/or post-translational modifications are required for regulating localization and enzymatic activity of Tet proteins. Fourth, the fact that 5hmC accumulates to a significant level in certain organs and tissues suggests that it may function as a bona fide epigenetic mark for the recruitment of specific “reader” proteins. Thus, identification of proteins that can specifically recognize 5hmC or other 5mC oxidation derivatives may reveal the function of these new epigenetic marks. Fifth, the observation that Tet proteins are capable of sequentially oxidizing 5mC/5hmC/5fC suggests that they can use different substrates. To elucidate substrate recognition and catalysis mechanisms, high-resolution cocrystallization structures of Tet proteins and their substrates are needed.
In addition to biochemical and genomic studies, a significant advance in understanding the biological functions of Tet proteins is the generation of mouse models for all three Tet genes. Given their unique expression patterns, it is anticipated that further functional studies of single and combinatorial knockout mice will uncover distinct as well as overlapping functions of different Tet proteins in the normal development of various somatic tissues and germ cells. Finally, given that the human TET2 gene is frequently mutated in a variety of hematopoietic neoplasms, integrative genome-wide analysis aimed at delineating direct targets of Tet2 during normal hematopoiesis will provide mechanistic insights into the role of Tet2 in myeloid malignancies. In summary, the discovery of the Tet family of DNA hydroxylases and related oxidation derivatives highlights the dynamic nature of epigenetic modification of DNA and suggests that DNA methylation and demethylation may play a critical role in diverse biological processes. If the exciting progress in the past 2 years is any indication, answers to the questions raised above will not be too far away.
Acknowledgments
We thank Susan Wu for critical reading of the manuscript. We also thank reviewers for their constructive comments and suggestions. We apologize to colleagues whose work cannot be cited owing to space constrains. This work was supported by NIH grants GM68804 and U01DK089565 (to Y.Z.). H.W. is supported by a Jane Coffin Childs post-doctoral fellowship. Y.Z. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
-
↵5 Corresponding author.
E-mail yi_zhang{at}med.unc.edu.
-
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.179184.111.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








