




Abstract
AKT, a phospholipid binding-serine/threonine kinase, is a key component of the phosphoinositide 3-kinase (PI3K) cell survival signaling pathway that is aberrantly activated in many human cancers. Many attempts have been made to inhibit AKT; however, selectivity remains to be achieved. We have developed a novel strategy to inhibit AKT through targeting the pleckstrin homology (PH) domain. Using in silico library screening and interactive molecular docking, we have identified a novel class of non-lipid based compounds that bind selectively to the PH domain of AKT, with “in silico” calculated KD values ranging from 0.8 μM to 3.0 μM. In order to determine the selectivity of these compounds for AKT, we used surface plasmon resonance to measure the binding characteristics of the compounds to the PH domains of AKT1, insulin receptor substrate-1 (IRS1) and 3-phosphoinositide-dependent protein kinase 1 (PDK1). There was excellent correlation of predicted in silico and measured in vitro KDs for binding to the PH domain of AKT which were in the range 0.4 to 3.6 μM. Some of the compounds exhibited PH domain binding selectivity for AKT compared to IRS1 and PDK1. The compounds also inhibited AKT in cells, induced apoptosis and inhibited cancer cell proliferation. In vivo, the lead compound failed to achieve blood concentrations required to inhibit AKT in cells, most likely due to rapid metabolism and elimination, and did not show anti-tumor activity. These results show these compounds as the first small molecules selectively targeting the PH domain of AKT.
Keywords: AKT, pleckstrin homology domain, inhibitor, PDK1, IRS1, drug design
Introduction
The phosphoinositide 3-kinase (PI3K) pathway is critical to many aspects of cancer cell function including growth, survival, differentiation, and invasion (1,2). PI3K phosphorylates membrane phosphatidylinositol–4,5-bisphosphate (PtdIns(4,5)P2) to give the trisphosphate PtdIns(3,4,5)P3, which binds to lipid binding domains of downstream targets, recruiting them to the membrane. PI3K signaling is activated by many growth factors and regulators of cell proliferation while the tumor suppressor PTEN (phosphatase and tensin homolog deleted in chromosome ten), a dual specificity tyrosine-threonine/PI3-phosphatase (3,4), prevents the accumulation of PtdIns(3,4,5)P3 and, thus, attenuates PI3K signaling (5). Constitutive activation of the pathway through mutation, amplification and rearrangement occurs frequently in human cancer and is associated with aggressive tumor growth, increased metastasis, and resistance to therapy (1,2). Thus, the PI3K pathway is an attractive target for cancer drug discovery that is actively being pursued by many pharmaceutical and academic groups.
The primary downstream mediator of the effects of PI3K is AKT (or protein kinase B), which binds PtdIns(3,4,5)P3 through its N-terminal pleckstrin homology (PH) domain. AKT is a 56 kDa member of the AGC serine/threonine kinase family (6). There are three known mammalian isoforms, AKT1/α, AKT2/β and AKT3/γ, which share a high degree sequence homology in their catalytic and N-terminal PH domains, but differ in the linker between the domains and the C-terminal extension (7). AKT1 and AKT2 are ubiquitously expressed whereas AKT3 is found predominantly in brain, heart, and kidney (8). Following PH domain membrane recruitment, AKTs are phosphorylated on conserved Thr308 in the activation loop and Ser473 in the C-terminal extension (9) resulting in activation of AKT kinase activity (7,10). Phosphorylation of both residues is required for AKT activation (11). The kinase responsible for Thr308 phosphorylation is the constitutively active 3-phosphoinositide-dependent kinase 1 (PDK1) (12) while Ser473 can be phosphorylated by integrin-linked kinase (ILK) (13), the mTOR rictor complex (14), PKCε (15) and by auto-phosphorylation (16). Phosphorylated AKT is independent of phospholipid activation and detaches from the plasma membrane translocating to the cytoplasm and the nucleus (17). The full activity of AKT in promoting cell survival relies on phosphorylation of a battery of targets to either prevent the expression of death genes or to induce cell survival (18). Promoters of apoptosis that are inhibited by AKT are the forkhead transcription factor family members (FKHR, FHHRL1 and AFX) (19), the pro-apoptotic Bcl-2 family member Bad, the apoptosis signaling kinase-1 (ASK1) that transduces stress signals to the JNK and p38 MAP kinase pathways (20), and procaspase-9, the initiator of the caspase cell death cascade (21). Targets that AKT activates to promote cell survival are NF-κB (22) and the cyclic AMP response element binding protein (CREB) (23). AKT also phosphorylates p70S6kinase (24) and GSK3β (25) contributing to cyclin D accumulation of cell cycle entry (26). Finally, AKT phosphorylates and activates mTOR/FRAP to increase hypoxia inducible factor- 1 α (HIF1α ), a mediator of VEGF production and angiogenesis (27).
The PH domain is a ~100 to 120 amino acid modular fold found in over 250 human proteins (28). PH domains have few critically conserved amino acids, but show remarkable conservation of three-dimensional structure. Crystal structures and nuclear magnetic resonance structures of several PH domains show a highly conserved three-dimensional organization although sequence identities are only 7% to 23%. The core of each PH domain consists of a β-barrel of seven anti-parallel β-strands and a C-terminal amphipathic α-helix. PH domains can bind to Gβγ subunits of heterotrimeric G proteins (29,30), to certain phosphotyrosine peptides, polyproline sequences, and phosphoinositides (PtdIns). A majority of PH domain members bind PtdIns weakly and non-specifically but a subclass of approximately 40 PH domain proteins shows high affinity for PtdIns. These PtdIns-binding PH domain proteins are important components of signal transduction pathways that regulate cancer cell growth and survival (31). PtdIns binding PH domains are classified according to their binding specificity based on conserved positively charged residues in the phosphatidylinositol phosphate (PtdInsP) binding pocket and have KDs in the range 1–5μM (32). Group 1 PH domains specifically recognize PtdIns(3,4,5)P3. Group 2 PH domains bind PtdIns(4,5)P2 and also interact with other phosphoinositides, but since PtdIns(4,5)P2 is more abundant than 3-phosphorylated phosphoinositides, PH domains in Group 2 are regulated by PtdIns(4,5)P2. Group 3 PH domains recognize PtdIns(3,4)P2 and PtdIns(3,4,5)P3. Group 4 PH domains have low affinity for PtdInsP binding. Group 2 PH domains mediate the effects of PtdIns(4,5)P2 on membrane trafficking and plasma membrane-cytoskeleton linkages while Group 1 and Group 3 domains mediate the effects of PtdIns(3,4,5)P3 on cell signaling pathways that regulate cell growth and survival (2). AKT has a Group 3 PH domain. The crystal structure of the PH domain of AKT1 bound to the inositol head group of PtdIns(3,4,5)P3, that is, myo-inositol(1,3,4,5)P4, has been determined and the mode of binding elucidated (32). Recently an activating mutation (Glu17 into Lys17) has been reported in the PH domain of AKT at low frequency in human tumos (33).
There have been attempts to develop selective inhibitors of the AKT kinase ATP catalytic site as potential anti-cancer drugs but a major problem has been toxicity, possibly associated with a lack of selectivity for other members of the AGC serine/threonine kinase family (34,35). We have previously reported that a PtdIns analog, D-3-deoxyphosphatidylinositol ether lipid (DPIEL, PX-316), binds selectively to the PH domain of AKT1, preventing the protein translocation and activation at the plasma membrane (36). In mice xenograft models of cancer, PX-316 inhibits tumor AKT and has anti-tumor activity (36,37). This work was the point of departure for a cancer drug discovery effort based on the x-ray crystal structure of the AKT1 PH domain that uses in silico library screening, docking, structure-based design, synthesis, testing, and iterative refinement to develop novel inhibitors of AKT.
Materials and Methods
Pharmacophore design, in silico screening, and interactive docking
The high-resolution crystal structure of the isolated PH domain of human AKT1 in a complex with the head group of Ins(1,3,4,5)P4 (32) was utilized to define a pharmacophore pocket for screening a 2,000 molecule database (National Cancer Institute) using Unity in Sybyl (version 7.2; Tripos Inc., St Louis, MO). The pharmacophore pocket included all the residues of the AKT1 crystal structure within 5Å of the Ins(1,3,4,5)P4 binding site: Lys14, Arg15, Gly16, Glu17, Tyr18, Ile19, Lys20, Thr21, Arg23, Pro24, Arg25, Lys39, Pro51, Leu52, Asn53, Asn54, Phe55, Gln79, Ile84, Glu85, Arg86 and Phe88. The program operates by assigning attributes to various atoms on the ligand or protein binding site. The defined pharmacophore pocket was used to search virtual chemical databases and candidate compound “hits” were identified. The FlexX docking algorithm in Sybyl V.7.2 was utilized for the docking of these compounds into the AKT1 PH domain active site. FlexX produces 30 different docking orientations (poses) of the ligand within the active site. Various docking orientations were analyzed on the basis of FlexX scores, G-score, and X-score. The scores are similar to interaction energy, and the more negative the value, the better the interaction. The predicted KD is calculated by pKD = 10 exp(−Xscore) (38). In order to investigate the possibility of specific binding of the identified small molecules at the AKT1 PH domain using in silico methods, known crystal structures of the IRS1 PH domain (IRS1, PDB:1QQG) (39) and of the PDK1 PH domain (PDK1, PDB:1W1D, 1W1G) (40) were also used for docking studies similar to those described above.
Synthetic Procedures
Details of the syntheses and characterizations of the compounds used herein are located in the Supplemental Data Section.
Expression and purification of recombinant PH domains
Recombinant mouse AKT1 PH domain amino acids 1–111 (UBI/Millipore, Charlottesville, VA), human AKT1 PH domain amino acids 1–111 (Origene NM005163.2), human IRS1 PH domain amino acids 12–133 (Invitrogen, #IOH29016) and human PDK1 PH domain amino acids 407–549 (Origene, NM002613.3) were cloned by PCR into EcoRI/XhoI sites in pGEX-4T1 inducible bacterial expression plasmid (GeneStorm, InVitrogen, Carlsbad CA) transformed into BL21(DE3) E. Coli.
Surface plasmon resonance (SPR) spectroscopy binding assays
All interaction analyses were performed with a Biacore 2000, Biacore 2000 Control Software v3.2, and BIAevaluation v4.1 analysis software (Biacore, Piscataway, NJ). The PH domain GST-fusion proteins (AKT1, IRS1, and PDK1) were immobilized on a CM5 Sensorchip (Biacore BR-1000–12) using Biacore’s Amine Coupling Kit (Biacore BR-1000–50) to a level of 10,000 Response units (RUs). Small molecule analytes at concentrations ranging from one tenth to ten times the predicted KD were injected at a high flow rate (30μL/min). Dimethylsulfoxide (DMSO) concentrations in all samples and running buffer were 1% (v/v) or less.
ELISA competitive binding assay
A 96-well Maxisorb plate (Nunc, Rochester, NY) was coated with 1μG/100μl L-α-phosphatidylinositol(3,4,5)P3 (Biomol, Plymouth Meeting, PA). The proteins (purified GST-PH domains) were incubated with the drugs for 4 hours in 0.2 M carbonate buffer pH 9.4. The proteins mixed with increasing concentration of drugs were then added to the 96-well plate and incubated overnight at 4ºC. The plate was washed 4 times with phosphate buffered 0.9% NaCl (PBS), blocked with 3% bovine serum albumin (BSA) in PBS and 0.01% Tween for 1 hour, washed again 4 times with PBS and mouse monoclonal anti-glutathione-S-transferase antibody (Pierce) in 3% BSA (1: 2000) was added for 1 hr at room temperature shaking. The plate was washed 4 times with PBS and an anti-mouse IgG horseradish peroxidase coupled antibody (dilution 1: 2000 in 3 % BSA) was added for 1 hr. Following 4 washes with PBS, 2,2′-azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABST, from Pierce) was added and the reaction was allowed to develop for 30 min. A stop solution of 1% sodium dodecyl sulfate was then added and the plate was read at 405 nm in a plate reader (μQuant Bio Tek Instruments Inc., Winooski, VT).
Cell culture and compound treatments
Human HT-29 colon cancer cells and NIH3T3 mouse fibroblast cancer cells were obtained from the American Type Culture Collection (Rockville, MD). Cells and maintained in bulk culture in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 4.5 g/l glucose, 100 U/ml penicillin and 100 μg/ml streptomycin in a 5% CO2 atmosphere. Cells were passaged using 0.25% trypsin and 0.02% EDTA. Cells were confirmed to be mycoplasma free by testing them with an ELISA kit (Roche-Boehringer Mannheim, Indianapolis, IN). The compounds were dissolved in DMSO at a stock concentration of 10 mM and then added at different concentrations directly into the culture media of the cells.
Inhibition of phospho-Ser473AKT, phospho-Ser241PDK1 and phospho-(pan)-PKC by Western analysis
Inhibition of the phosphorylation of AKT,1 PDK1 or PKC was measured by Western blotting as described previously (36) using rabbit polyclonal antibodies to phospho -Ser473-AKT, pan-phospho-PKCs, and phosho-Ser241-PDK1 (New England Biolabs/Cell Signaling Technology Inc., Danvers, MA). Bands corresponding to phospho-AKT, phospho-PKC or phospho-PDK1 were quantified using Eagle Eye software (BioRad, Richmond, CA) and Kodak X-Omat™ Blue XB (NEN™, Life Science Products). β-actin was used as a loading control in all Western blots.
Cell cytotoxicity assay
Cell growth inhibition was determined using a micro-cytoxicity assay. Cells were plated in 96-well micro-cytoxicity at 5,000–10,000 cells per well (depending on cell doubling time) and grown for 7 days. Compounds dissolved in DMSO were added directly to the media, at various concentrations ranging from 1 to 50 μM. The endpoint was spectrophotometric determination of the protein content of each well using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. A concentration-response relationship at two or more concentration levels was used to obtain an IC50. for the compound.
Cell Apoptosis assay
Apotposis was measured as described previously in reference 41. Briefly, HT-29 cells were grown to 70–75% confluency in 6-well tissue culture plates. Cells were treated with the compounds for 24 hours. To measure apoptosis, 10 μl of cells were mixed with ethidium bromide and acridine orange solution (100 μg/ml each in DMEM) and visualized by immunofluorescence for morphological changes. A minimum of 200 cells was counted and the percentage of apoptotic cells determined.
Anti-tumor activity
Approximately 1×107 HT-29 colon cancer cells in log phase growth were injected subcutaneously (sc) in 0.1 ml PBS into the flanks of female severe combined immunodeficient (scid ) mice. When the tumors reached volumes between 100 and 170 mm3, the mice were stratified into groups of 8 animals having approximately equal mean tumor volumes and administration of compound 1 suspended in 0.2 ml 25% dimethylsulfoxide in 20% pharmaceutical grade hydroxypropyl-β-cyclodextrin (Trappsol®, Cyclodextrin Technologies Development, High Springs, FL) in water was started at a dose of 250 mg/kg per day per os (po) daily for 5 days. The animals were weighed weekly and tumor diameters measured twice weekly at right angles (dshort and dlong) with electronic calipers and converted to volume by the formula volume = (dshort)2 × (dlong)/2 (42). When the tumor volume reached 2,000 mm3 or became necrotic, the animals were euthanized.
Pharmacokinetic Studies
Male C57Bl/6 mice were administered compound 1 intraperitonealy (ip) or po at 250 mg/kg suspended in 0.2 ml 25% DMSO in 20% Trappsol®. The mice were sacrificed at various times, blood was collected into heparinized tubes and plasma was prepared. Plasma (0.2 ml) was immediately mixed with an equal volume of 0.25 M sodium phosphate buffer at pH 4.0 and extracted for 1 hr by inversion with 4 ml ethyl acetate. After centrifugation, 3.8 ml of the organic layer was removed and evaporated under N2 and the residue dried on a lyophilizer. Chromatographic separation was achieved with a Waters Symmetry C-18 3.9 × 150 mm column (Waters, Milford, PA) with a mobile phase of 0.1% trifluoroactetic acid in 60% methanol, at a flow rate of l/min with detection at 254 nm. For the assay, the sample residue was dissolved in 100μl mobile phase and centrifuged at 15,000 g for 5 min at 4°C. The limit of detection of the assay for all the compounds from 0.2 ml mouse plasma was 0.01 μg/ml.
Toxicity Studies
Compound 1 was administered at 250 mg/kg per day by oral gavage (po) daily for 5 days to female scid mice. The mice were sacrificed 24 hr after the last dose and changes in body weight from the start of the study, blood lymphocyte, neutrophil, red blood cell and platelet counts, and aspartate amino transferase (AST) and amino alanine transferase (ALT) were measured.
Pharmacodynamic Studies
1×107 HT-29 colon cancer cells were injected sc into the flanks of female scid mice and allowed to grow to approximately 300 mm3. Mice received a single ip dose of compound 1 of 250 mg/kg in 25% DMSO in 20% Trappsol® in water. Mice were killed at various times and tumors removed and immediately frozen in liquid N2. Tissues were homogenized in 50mM HEPES buffer, pH 7.5, 50mM NaCl, 1% Nonidet® P40 and 0.25 % sodium deoxycholate, and Western blotting performed using anti-phospho-Ser473-AKT and anti-AKT antibodies. AKT activation was expressed as the ratio of phospho-Ser473-AKT to total AKT.
RESULTS
Discovery of lead molecule 1 (NSC 348900) and its binding to the AKT1 PH domain
PtdIns(1,3,4,5)P4 and its reported interactions with the crystal structure of the AKT1 PH domain (32) provided the starting point for the identification and design of AKT1 PH domain small molecule inhibitors. A pharmacophore query search using the National Cancer Institute database led to the identification of several lead molecules. These lead molecules were docked and then ranked based on their docking scores. One of these molecules - NSC348900 (compound 1) exhibited good FlexX score and G-score values as summarized in Table I and was selected as a lead for future studies. The predicted binding affinity (KD) of compound 1 to the AKT1 PH domain was 1.2 μM, which was three times better than the lipid-based compound, DPIEL with a predicted KD of 4.0 μM (Table I). Figure 1A shows the predicted binding of compound 1 to the PH domain binding pocket of AKT1 and Figure 1B represents hydrogen bonding interactions that occur between compound 1 and the amino acid side chains, as well as the backbone of the AKT1 PH domain binding pocket. The sulfonamido group interacts with Arg86 through a hydrogen bond while a similar hydrogen bonding interaction is involved with the diazopyrazolyl group with Arg23. These two arginine residues are involved in the strong interaction with the phosphate head groups of the substrate PtdIns(1,3,4,5)P4 supporting the discovery of compound 1 as an AKT1 PH domain inhibitor. Other hydrogen bonds are also established between the backbones of Ile19 and Asn53 with the sulfonamide function of the compound.
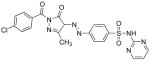
Table I. Structures and calculated properties of lead compound 1 (NSC 348900) and analogues (2–6).
| Compounds | AKT1 FlexX Score |
AKT1 G Score |
AKT1 cKD (μM) |
PDK1 FlexX Score |
PDK1 G Score |
PDK1 cKD (μM) |
IRS1 FlexX Score |
IRS1 G Score |
IRS1 cKD (μM) |
|
|---|---|---|---|---|---|---|---|---|---|---|

|
NS | NS | 4.0 | NS | NS | NS | NS | NS | NS | |
| 1 |
|
−31.0 | −96.9 | 1.2 | −17.4 | −109.0 | 1.74 | −16.0 | −128.0 | 1.99 |
| 2 |

|
−29.6 | −31.9 | 2.4 | −17.0 | −40.0 | 2.60 | −17.1 | −96.2 | 2.40 |
| 3 |

|
−28.2 | −99.5 | 1.2 | −17.1 | −103.4 | 1.70 | −14.8 | −79.7 | 10.70 |
| 4 |
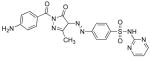
|
−29.1 | −71.9 | 3.0 | −17.5 | −88.6 | 2.20 | −17.9 | −145.5 | 1.80 |
| 5 |
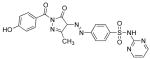
|
−33.0 | −120.6 | 1.3 | −20.1 | −137.1 | 2.40 | −14.6 | −90.1 | 10.70 |
| 6 |
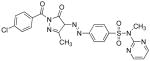
|
−24.3 | −132.0 | 0.85 | −21.0 | −109.1 | 1.45 | −14.5 | −140.6 | 0.52 |
NS: for not shown
Figure 1. Interactions of compound 1 with the human AKT1 PH domain.

Panel A is a schematic representation of the interaction of compound 1 (NSC 348900, 4-[1-(4-Chlorobenzoyl)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-4-ylazo]-N-pyrimidin-2-yl-benzenesulfonamide (C21H16ClN7O4S) with amino acid residues of the AKT1 PH domain (Arg86, Asn53, Arg23 and Ile19). Panel B represents the docking pose of compound 1 with the AKT1 PH domain. The AKT1 PH domain is colored red and residues Arg23, Arg25 and Arg86 colored by atom type. Compound 1 is represented as capped stick and colored by atom type. Hydrogen bonding interactions are displayed as dotted lines. The sulfamido group interacts with Arg86 through a hydrogen bonding interaction while similar hydrogen bonds are involved in the interaction of diazopyrazolyl group with Arg23. These two arginines are intimately involved in the interaction with the phosphate head groups of the substrate phosphoinositol-1,3,4,5-tetrakisphosphate. Panels C and D represent the binding mode of compound 1 in the binding pocket of the PH domain of PDK1 with the amino acids involved in the binding pocket (Panel D). Note that compound 1 exhibits the reverse binding pose in the PDK PH domain similar to compound 2 in the PH domain of AKT1. In the supplementary Figure 1, panel A and B represent the binding mode of compound 1 in the binding pocket of the PH domain of IRS1.
Rational Design, Chemical Synthesis, and Docking Studies of Compounds (2–6)
In order to test whether modifications of compound 1 could affect the binding properties and activity against AKT1, compound 1 and five structural analogs (compounds 2 to 6) with varying chemical properties were synthesized (Scheme 1). Diazotization of sulfadiazine (7) with sodium nitrite under acidic conditions, followed by treatment of the diazonium salt with ethyl acetoacetate and sodium acetate, gave β-ketoester 8 in 95% yield. Condensation of 8 with different benzoylhydrazides in glacial acetic acid at 100 °C produced NSC 348900 (1) and analogs 2–5 in yields ranging from 19%–71%. Compound 6 was prepared by treatment of compound 1 with sodium hydride and methyl iodide in THF (reaction not shown). Details of these syntheses and spectroscopic data for compounds 1–6 and 8 are given in the Supplemental Data Section. The structures and docking scores for these compounds are summarized in Table I. Analyses of the docking poses of these compounds in the PH domain of AKT1 revealed different docking orientations between compounds 1, 3 and 5 as compared to compounds 2, 4 and 6. Since there are only small changes in the structures of these compounds, it is expected that these differences in docking orientations are due to limitations of the FlexX docking program (for flexible compounds). It was expected that compounds 1 – 6 would exhibit similar binding to the AKT1 PH domain.
Scheme 1.

Synthesis of Compounds 1–6.
The sequence alignments of critical binding residues in the PH domains of the three human isoforms AKT1, AKT2 and AKT3 are identical (Table II) and thus indicate that compounds 1–6 should bind equally well to the three isoforms of AKT. KDs were also calculated for the binding of the compounds 1 to 6 to the PH domain of PDK1 and were found to be very similar to those for AKT1 (Table 1). The calculated KDs for the binding of compounds 1 to 6 to the PH domain of IRS1 showed greater variability with compound 6 having the greatest affinity and compounds 3 and 5 more than an order of magnitude lower affinity. Figures 1C and 1D represent the binding mode of compound 1 in the binding pocket of the PH domain of PDK1 (Supplemental Figures 1A and 1B show the binding mode of compound 1 in the binding pocket of the PH domain of IRS1). Compound 1 exhibits the reverse docking mode in IRS1 PH and PDK1 PH domains similar to compound 2 in the PH domain of AKT1.
Table II. Sequence alignments of the PH domain of AKT.
The PH domain binding residues are shown underlined in the sequence alignment below obtained using CLUSTAL W (1.82) multiple sequence alignment.

|
Measured Binding Affinity of Compound 1 and Analog Compounds 2–6
In order to validate the in silico data and the docking studies, binding assays using the Biacore SPR biosensor and an ELISA competitive binding assay were used to measure the binding affinity (KD) of the compounds to all three PH domains. Table III summarizes the results obtained from the Biacore SPR measurements, which correlated well with the predicted KD values for the compounds for each PH domain. Representative saturation curves as well dose response curves are shown in Figure 2 for compounds 1 and 2 to the PH domain of AKT1 (panel A) and to the PH domain of IRS-1 (panel B). Compounds 1 and 2, which modeling suggest bind in a reverse binding pose in the PH domain binding pockets of the three different PH domains, also exhibited very different biosensor binding curves. An ELISA competitive binding assay was conducted using the PH domains of AKT1 and IRS1 with compounds 1 (Figure 3 panel A) and 2 (Figure 3, panel B). The ELISA gave binding IC50s with AKT1 for compound 1 and 2 of 0.08 μM, and with IRS1 for compound 1 of 1.0 μM, and compound 2 of > 100 μM. These IC50s were similar to ones obtained using the Biacore.
Table III. Selectivity for other PH domains.
Binding affinities were measured by surface plasmon resonance spectroscopy as described in the Materials and Methods Section and are referred as mKD (μM) for measured.
| Compounds | AKT1 PH mKD (μM) | IRS1 PH mKD (μM) | PDK1 PH mKD (μM) |
|---|---|---|---|
| PtdIns(3,4,5)P3 | 3.08±0.49 | ND | ND |
| DPIEL | 5.04±0.48 | 31.56±8.49 | NB |
| 1 | 0.37±0.04 | 0.39±0.01 | 31.28±9.54 |
| 2 | 3.66±0.03 | NB | 0.17±0.10 |
| 3 | 1.37±0.25 | NB | 3.57±0.96 |
| 4 | 0.51±0.06 | 0.14±0.02 | NB |
| 5 | 1.35±0.02 | 1.74±0.41 | 0.42±0.17 |
| 6 | 1.62±0.02 | NB | 0.98±0.48 |
NB = no measurable binding, ND = not determined.
Figure 2. Binding of the compounds (1–2) to the PH domain of AKT1 and IRS1.

The proteins were immobilized on a CM5 Sensorchip as described in the Materials and Methods section. The drugs were injected over the surface at the indicated concentrations and binding to the proteins was measured by surface plasmon resonance. Panel A represents the binding curve of compound 1 (left) and compound 2 (right) to the PH domain of AKT and Panel B shows the binding curves of compound 1 (left) and 2 (right) binding to the PH domain of IRS1. These results show an overlay plot of typical sensorgrams obtained with increasing concentrations of compound 1 or 2 as shown by the arrows (Panel A)
Figure 3. ELISA competitive binding assay.

Competition binding curves for compounds 1 (Panel A) and 2 (Panel B) respectively to the (□) AKT PH domain and (●) IRS1 PH domain and Values are the mean of 3 determinations and bars are SD with **,p< 0.05. Compound 1 binds the PH domains of AKT1 and IRS1 in a similar manner. Compound 2 binds only the PH domain of AKT1.
Finally, consistent with the docking studies, compounds 1, 4 and 5 exhibited low KD while compounds 2, 3 and 6 do not show any binding to IRS-1 PH domain as measured by Biacore SPR. However, compounds 4 and 6 did not bind the PH domain of PDK1 while the calculated KD was predicted to be 2.2 and 1.4 respectively. Taken together, these data suggest that the structural modifications in compound 1 have altered the binding positions of the compounds in the AKT1 PH domain as well as modified their specificity against IRS1 or PDK1 PH domains. Overall, the predicted binding affinities correlated well with measured binding constants for AKT1 and PDK1 PH domains but were less accurate for IRS-1 PH domain.
Biological activities of compounds 1–6 in cancer cells
Next, the ability of the compounds (1–6) to inhibit phospho-Ser473AKT was measured in cancer cells (Table IV). All except compound 3, the most lipophilic of the compounds, inhibited phospho- Ser473AKT with IC50’s ranging from 2 to 10 fold higher than required to bind to the AKT1 PH domain. Figure 4 shows typical Western blots obtained for the compounds in HT-29 colon cancer cells (Panels A) and with concentration responses for compound 1 (Panel C) and compound 2 (Panel D). AKT phosphorylation was decreased in a concentration-dependent manner by increasing amounts of compounds 1 and 2 (Panels C and D respectively). The phosphorylation of PDK and downstream target PKC were also decreased upon treatment of the cells with compound 1 (Panel C). IRS1 phosphorylation could not be detected in these cells (data not shown). Compound 2 mainly inhibited Akt phosphorylation with no effect on the phosphorylation of PDK or PKC. Cytotoxicity for compounds 1, 2 and 3 was in the same range for inhibition of cell phospho-Ser473AKT while coumpounds 4 and 5 showed no cytotoxicity (Table IV). Apoptosis was also measured and correlated well with Akt inhibition in HT-29 (Figure 4, Panel B). Compounds 2 and 6 induced 60 to 50% apoptosis at 20μM and both compounds inhibited AKT as well as downstream targets such as GSK3 phosphorylation (Panel A).
Table IV. Biological Properties of Compound 1 and analogs.
For each of the analogs phospho-Ser473 AKT inhibition was measured in either mouse NIH3T3, or human HT-29 colon cancer cells. Cytotoxicity was measured in HT-29 cells. Metabolic stability was measured by incubating with HT-29 cells at the maximum concentration in DMEM at 37°C. The apparent permeability (nm/sec) in Caco-2 and MDCK cells was obtained using the QikProp software (Schrodinger Inc., San Diego, CA).
| Compound | AKT inhibition (IC50 μM) |
Cyto-toxicity (IC50 μM) |
LogP | Metabolism half life (min) | Solubility (μM) |
Permeability (nm/sec) Caco2, MDCK |
|
|---|---|---|---|---|---|---|---|
| NIH3T3 | HT-29 | ||||||
| 1 | 4 | 13 | 24 | 2.1 | 62 | 17.9 | 90, 91 |
| 2 | 11 | 20 | 14 | 1.9 | 62 | 28.3 | 83, 34 |
| 3 | >20 | >20 | 25 | 3.2 | 91 | 28.6 | 95, 39 |
| 4 | ND | >20 | NI | 1.2 | >480 | 12.9 | 23, 8 |
| 5 | 5 | >20 | NI | 1.5 | 138 | 13.1 | 185, 200 |
| 6 | 3 | 5 | ND | 1.9 | ND | <0.1 | 14, 5 |
Values ranging <25 for poor and >500 for great permeability. NI=not inhibitory for IC50 >100 μM; ND= for not determined.
Figure 4. Inhibition of AKT in cancer cells.


In Panel A, HT-29 colon cancer cells were treated with compounds 1–6, at 20μM for 2 hr and stimulated with 50 ng/nl EGF for 30 min. Akt activity was measured by Western blotting using anti-phosphoSer437 AKT antibody. All other downstream targets of AKT were detected also by Western blotting using specific anti-phospho antibodies. β-actin was used as a loading control. Note that compounds 2 and 6 inhibit AKT phosphorylation and downstream GSK3 phosphorylation. Panel B shows apoptosis measured as described in the Material and Methods Section. Note that both compound 2 and 6 at 20μM induce significant apoptosis significantly as compared to controls. Values are the mean of 3 determinations and bars are SD with ** p< 0.05 and ***,p<0.001. Compound 1 (Panel C) or its analog 2 (Panel D) were also tested at the concentrations shown for 2 hr, and in HT-29 cells stimulated with 50 ng/nl EGF for 30 min. Akt activity was measured by Western blotting using anti-phosphoSer437 AKT antibody, PDK activity by anti-phosphoSer241 PDK antibody as well as downstream target PKC using pan-phosphoPKC antibodies. β-actin was used as a loading control.
In vivo effects of the AKT1 PH domain inhibitor
Absorption, metabolism, distribution and elimination (ADME) properties of compound 1 (NSC 348900) were predicted using the QikProp software (Schrodinger Inc., San Diego, CA). Metabolism predictions suggested that the azo- (-N=N-) linkage in the compounds may be susceptible to metabolic reduction. The stability of the compounds in cell culture conditions was measured and showed relatively rapid breakdown for compounds 1, 2, 3 and 5 with half lives of 1 to 2 hr, while compound 4 showed no breakdown over the time period studied (Table IV). Compound 6 was too insoluble to obtain any data.
Compound 1 was chosen for in vivo evaluation as the most promising and potent AKT PH domain binding compound. The compound is very insoluble and thus was administered as a slurry in 25% DMSO 20% Trappsol®. Preliminary studies showed no toxicity as a single dose up to 250 mg/kg which was the maximum dose that could practically be administered ip. Five daily doses at 250 mg/kg ip gave a moderate neutropenia but no other sign of toxicity with no change in body weight, blood lymphocyte, red blood cell and platelet count, or aspartate amino transferase (AST) or amino alanine transferase (ALT). Pharmacokinetic studies of a single dose of compound 1 of 250 mg/kg showed a peak concentration of 1.4 μM for ip administration and 0.6 μM for oral administration with a relative area under the plasma concentration time curve for oral compared to ip administration of 53.0% (Figure 5A). The failure to achieve high plasma concentrations and the relatively rapid elimination of parent compound over 24 hr despite the very large doses given, suggest rapid metabolism or elimination. Concentrations required to inhibit AKT based on the cell studies of around 4 to 13 μM were not achieved. Anti-tumor studies showed no activity of compound 1 given orally daily for 5 days at 250 mg/kg against HT-29 colon cancer (Figure 5B). There was a small inhibition of tumor phospho-Ser473AKT 4 hr after a single dose of compound 1 but no inhibition at 24 hr (Figure 5C). Unexpectedly, at 24 hr there was a significant decrease in total AKT compared to the actin loading control. Taken together, the results suggest that the limited solubility of compound 1, and metabolism or elimination, limit the plasma concentrations that can be achieved, thus, preventing effective inhibition of AKT and possible anti-tumor activity. Despite no clear anti-tumor activity, given that compound 1 can inhibit AKT phosphorylation, it can be hypothesized that tumor cells may be sensitized for and be susceptible to chemotherapy and/or radiation treatment.
Figure 5. In vivo activity of compound 1.

A, Pharmacokinetics of compound 1 in mice. Female scid mice were administered compound 1 at a dose 250 mg/kg either (●) intraperitonealy or (○) po by oral gavage and plasma concentrations measured. Values are the means of 3 mice and bars are S.E. B, Antitumor activity in female scid mice with HT-29 colon cancer xenografts treated orally daily for 5 days (shown by the arrows) with (●) vehicle alone or (□) compound 1 at 250 mg/kg daily. Values are the means of 10 mice and bars are S.E. C. Effect on tumor phospho-AKT in female scid mice with HT-29 colon cancer xenografts treated orally with (open bar) vehicle alone or (filled bars) compound 1 at 250 mg/kg. Tumors were removed at various times for Western blotting. Values are the mean of 4 mice and bars are S.E. *p< 0.05,** p< 0.01.
Discussion
Targeting AKT for the treatment of cancer has remained challenging (reviewed in reference 43). Several ATP site inhibitors of Akt are relatively toxic compounds, probably because other serine/threonine kinses are being inhibited (43). On the other hand the lipid based PH domain inhibitors such DPIEL and Perifosine are relatively non toxic compounds (37,44). Thus we believe that inhibiting the PH domain could offer a better therapeutic approach than the ATP site inhibitors. We and others have developed lipid-based inhibitors of AKT designed to bind the PH domain of AKT (36,37,45,46). These compounds were shown to inhibit AKT translocation, phosphorylation and to induce apoptosis in cancer cells (36, 47). However, potency and lack of oral bioavailability and the main problems associated in the further development of this type of compound. Thus, we sought to develop novel chemical small molecules capable of binding the PH domain of AKT. In this study, we identified a non-lipid based compound (NSC 348900) and several analog compounds have been synthesized and tested in vitro and in vivo for their ability to bind the PH domain of AKT. Some of the compounds exhibited selectivity for the PH domain of AKT compared to IRS1 and PDK1 PH domains. These compounds showed some activity in cells model but did not show strong anti-tumor activity in vivo model of cancer.
The docking models developed for AKT PH domain inhibition used three different docking scores – FlexX score, G-score and X-score to evaluate the interaction of the compounds with the targeted protein. These models were successful in predicting Kd values for binding to the AKT1 PH domain for the different compounds compared to KDs measured using SPR technology.. However, there were apparent inconsistencies in the docking model of compounds 1–6 in the PH domain of IRS1. This might be because the stability and the specificity of IRS1 PH domain binding to phosphoinositides is different compared to the PH domain of AKT. Competitive binding of phosphoinositides to IRS1 PH domain indicate that phosphorylation at the 5 position on the myo-inositol ring contributes to the affinity and specificity since PtdIns(3,4,5)P3 and PtdIns(4,5)P2 bind to the PH domain with greater affinity than PtdIns(3,4)P2 and PtdIns(4)P. Binding complexes of phosphoinositides to isolated PH domains have been reported to be unstable (39–40). Second and more importantly, the docking orientation of the compounds may explain the differences between modeling and in vitro results using SPR measurements. As shown in Figure 1, the docking orientations of compound 1 in AKT1 PH domain and IRS1 PH domain are different (flipped). In case of the AKT1 PH domain, the chlorophenyl ring (R group) is exposed to the water pocket and does not interact extensively with the PH domain residues. In case of the IRS1 PH domain, the R group interacts with the active site residues of the PH domain pocket. Hence, the changes in the R substituents showed significant effect on binding to IRS-1 PH domain. We show for the first time that some of the compounds were also capable of binding to the PH domain of PDK1. We found that the compounds exhibit lower activity against AKT in cells that against the expressed PH domain. Increasing the bioavailability of such compounds may lead inhibition of AKT activity. A recent study has reported that PDK1 binds in a complex with AKT potentially via the PH domain of Akt, regulating the kinase’s activation (48). This may interfere with the ability of the compounds to bind to the PH domain of AKT. Finally, although these compounds were identified and designed to bind specifically the PH domain of AKT, one cannot rule out their possible effects on other kinases involved in the pathway.
Metabolism predictions on compound 1 suggested that the azo (-N=N-) linkage is metabolically unstable. These results may explain anti-tumor studies of the most specific and potent of the compounds, 1, which showed no activity against HT-29 colon cancer xenografts in mice. There was no inhibition of tumor phospho-Ser473 AKT most likely due to a failure to achieve sufficient plasma concentrations because of rapid metabolism or elimination despite the lack of toxicity and the large doses that could be administered. Taken together, the results suggest that inadequate solubility and pharmacokinetic properties may limit the ability of the azo sulfonamides developed in this study to inhibit AKT in vivo. We have, however, previously reported that the lipid compound D-3-deoxyphosphatidylinositol ether lipid (DPIEL, PX-316) that binds to the PH domain of AKT1, when administered to mice inhibits tumor AKT activity and exhibits moderate anti-tumor activity (36,37). This was observed despite the fact that DPIEL does not have properties of a good drug molecule. Thus, while AKT PH domain inhibitors can work in vivo and exhibit anti-tumor activity the design of more metabolically stable and soluble analogs with improved drug-like properties should improve the activity of this new class of compounds.
In summary, we have identified using in silico library screening and interactive molecular docking a novel class of non-lipid based compounds that bind selectively to the PH domain of AKT. Calculated KDs compared favorably with measured KDs used surface plasmon resonance. Some of the compounds exhibited PH domain binding selectivity for AKT compared to IRS1 and PDK1. The compounds inhibited AKT in cells and inhibited cancer cell proliferation. The lead compound failed to achieve blood concentrations required to inhibit AKT in cells, most likely due to rapid metabolism and elimination, and did not show anti-tumor activity.
Acknowledgments
This work was supported by grants RO1 CA 061015 and P30 CA 23074 from the National Cancer Institute to GP and by grant ABRC #9010 from the Arizona Biomedical Research Commission to EJM. Also we wish to thank Dr. Hariprasad Vankayalapati for input into pharmacophore design and screening of in silico databases.
Abbreviation List
- ABTS
2,2′-Azinobis[3-ethylbenzothiazoline-6-sulfonic acid]diammonium salt
- ADME
absorption, metabolism, distribution, and elimination
- ALT
amino alanine transferase
- ASK1
apoptosis signaling kinase-1
- AST
aspartate amino transferase
- CREBP
cyclic AMP response element binding protein
- DMSO
dimethylsulfoxide
- DPIEL
deoxyphosphatidylinositol ether lipid
- HIF
hypoxia inducible factor
- ILK
integrin-linked kinase
- IRS1
insulin receptor substrate-1
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- mTOR
mammalian target of rapamycin
- PDK1
3-phosphoinositide-dependent protein kinase 1
- PH
pleckstrin homology
- PtdIns
phosphatidylinositol
- PI3K
phosphoinositide 3-kinase
- PTEN
phosphatase and tensin homolog deleted in chromosome ten
- SPR
surface plasmon resonance
- TLC
thin-layer chromatography
- VEGF
vascular endothelial growth factor
References
- 1.Workman P, Clarke PA, Guillard S, Raynaud FI. Drugging the PI3 kinome. Nature Biotechnology. 2006;24:794–6. doi: 10.1038/nbt0706-794. [DOI] [PubMed] [Google Scholar]
- 2.Hennessy BT, Smith DL, Ram PT, Lu Yling Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nature reviews Drug Discovery. 2004;4:988–04. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 3.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 4.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1998;95:15587–91. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantley L, Neel BG. New insights into tumor suppression: PTEN suppress tumor formation by restraining the phosphoinositide 3-kinase/AKT pathways. Proc Natl Acad Sci USA. 1999;96:4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marte BM, Downward J. PKB/AKT: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–8. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 7.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-AKT): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakatani K, Thompson DA, Barthel A, et al. Up-regulation of AKT3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J Biol Chem. 1999;274:21525–32. doi: 10.1074/jbc.274.31.21528. [DOI] [PubMed] [Google Scholar]
- 9.Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Develop. 1998;8:55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- 10.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/AKT. FEBS Lett. 2003;546:108–12. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 11.Scheid MP, Woodgett JR. PKb/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2:760–8. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- 12.Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7:261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 13.Delcommenne M, Tan C, Gray V, et al. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–6. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarbassov dos D, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of PKB by the mTor-rictor complex. Science. 2005;307:1098–01. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 15.Kawakami Y, Nishimoto H, Kitaura J, et al. Protein kinase CβII regulates AKT phosphorylation on Ser-473 in a cell type and stimulus specific fashion. J Biol Chem. 2004;279:47720–5. doi: 10.1074/jbc.M408797200. [DOI] [PubMed] [Google Scholar]
- 16.Yang ZZ, Tschopp O, Hemmings-Mieszczak M, et al. Protein kinase Bα/AKT1 regulates placental growth and fetal growth. J Biol Chem. 2003;278:32124–31. doi: 10.1074/jbc.M302847200. [DOI] [PubMed] [Google Scholar]
- 17.Meier R, Alessi DR, Cron P, Andjelkovic M, Hemmings BA. Mitogenic activation, phosphorylation, and nuclear translocation of protein kinase Bβ. J Biol Chem. 1997;272:30491–7. doi: 10.1074/jbc.272.48.30491. [DOI] [PubMed] [Google Scholar]
- 18.Nicholson KM, Anderson NG. The protein kinase B/AKT signaling pathway in human malignancy. Cell Sign. 2002;14:381–95. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 19.Datta SR, Dudek H, Tao X, et al. AKT phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 20.Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. AKT phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 22.Ozes ON, Mayo LD, Gustin JA, et al. NF-kappaB activation by tumour necrosis factor requires the AKT serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 23.Du K, Montminy M. CREB is a regulatory target for the protein kinase AKT/PKB. J Biol Chem. 1998;273:32377–9. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 24.Chung J, Grammer TC, Lemon KP, Kazlauskas A, Blenis J. PDGF- and insulin-dependent pp70S6k activation mediated by phosphatidylinositol-3-OH kinase. Nature. 1994;370:71–5. doi: 10.1038/370071a0. [DOI] [PubMed] [Google Scholar]
- 25.van Weeren PC, de Bruyn KM, de Vries-Smits AM, Van Lint J, Burgering BM. Essential role for protein kinase B(PKB) in insulin-induced glycogen synthase kinase 3 inactivation: characterization of dominant-negative mutant of PKB. J Biol Chem. 1998;273:13150–6. doi: 10.1074/jbc.273.21.13150. [DOI] [PubMed] [Google Scholar]
- 26.Kerbel RS, Yu J, Tran J, et al. Possible mechanisms of acquired resistance to anti-angiangenic drugs: implications for the use of combination therapy approaches. Cancer Metast Rev. 2001;20:79–86. doi: 10.1023/a:1013172910858. [DOI] [PubMed] [Google Scholar]
- 27.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1 alpha expression by the epidermal growth factor/phosphatidylinositol-3 kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–5. [PubMed] [Google Scholar]
- 28.Rebecchi MJ, Scarlata S. Pleckstrin homology domains: a common fold with diverse functions. Ann Rev Biophys Biomol Struct. 1998;27:503–28. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- 29.Touhara K, Inglese J, Pitcher JA, Shaw G, Lefkowitz RJ. Binding of G protein beta gamma-subunits to pleckstrin homology domains. J Biol Chem. 1994;269:10217–20. [PubMed] [Google Scholar]
- 30.Mahadevan D, Thank N, Singh J, et al. Structural studies of the PH domains of Dbl, SOs1, IRS-1 and βARK1 and their differential binding to Gβγ subunits. Biochem. 1995;34:9111–7. doi: 10.1021/bi00028a021. [DOI] [PubMed] [Google Scholar]
- 31.Czech MP. Pip2 and pip3: complex roles at the cell surface. Cell. 2000;100:603–6. doi: 10.1016/s0092-8674(00)80696-0. [DOI] [PubMed] [Google Scholar]
- 32.Thomas CC, Deak M, Alessi DR, van Aalten DM. High-resolution structure of the pleckstrin homology domain of protein kinase b/AKT bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol. 2002;12:1256–62. doi: 10.1016/s0960-9822(02)00972-7. [DOI] [PubMed] [Google Scholar]
- 33.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–44. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 34.Feldman R, Wu J, Polokoff M, et al. Novel small molecule inhibitors of 3′-phosphoinositide-dependent kinsase1 (PDK-1) Eur J Cancer Supp. 2004;2:77. [Google Scholar]
- 35.Giranda V, Luo Y, Li Q, et al. Novel ATP-competetive AKT inhibitors slow the progression of tumors in vivo. Eur J Cancer Supp. 2004;2:77. [Google Scholar]
- 36.Meuillet EJ, Mahadevan D, Vankayalapati H, et al. Specific inhibition of the AKT1 pleckstrin homology domain by D-3-deoxy-phosphatidyl-myo-inositol analogs. Mol Cancer Ther. 2003;2:389–99. [PubMed] [Google Scholar]
- 37.Meuillet EJ, Ihle N, Baker AF, et al. In vivo molecular pharmacology and antitumor activity of the targeted AKT inhibitor PX-316. Oncology Research. 2004;14:513–27. doi: 10.3727/0965040042380487. [DOI] [PubMed] [Google Scholar]
- 38.Wang R, Lai L, Wang S. Further Development and Validation of Empirical Scoring Functions for Structure-Based Binding Affinity Prediction. J Comput-Aided Mol Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 39.Dhe-Paganon S, Ottinger EA, Nolte RT, Eck MJ, Shoelson SE. Crystal structure of the pleckstrin homology-phosphotyrosine binding (PH-PTB) targeting region of insulin receptor substrate 1. Proc Natl Acad Sci. 1999;96:8378–83. doi: 10.1073/pnas.96.15.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komander D, Fairservice A, Deak M, et al. Structural Insights Into the Regulation of Pdk1 by Phosphoinositides and Inositol Phosphates. EMBO J. 2004;23:3918–28. doi: 10.1038/sj.emboj.7600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powell AA, LaRue JM, Batta AK, Martinez JD. Bile acid hydrophobicity is correlated with induction of apoptosis and/or growth arrest in HCT116 cells. Biochem J. 2001;356:481–486. doi: 10.1042/0264-6021:3560481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paine-Murrieta GD, Taylor CW, Curtis RA, et al. Human tumor models in the severe combined immune deficient scid mouse. Cancer Chemother Pharmacol. 1997;40:209–214. doi: 10.1007/s002800050648. [DOI] [PubMed] [Google Scholar]
- 43.Kumar CC, Madison V. AKT crystal structure and AKT-specific inhibitors. Oncogene. 2005;24(50):7493–501. doi: 10.1038/sj.onc.1209087. [DOI] [PubMed] [Google Scholar]
- 44.Catley L, Hideshima T, Chauhan D, et al. Alkyl phospholipids perifosine induces myeloid hyperplasia in a murin myeloma model. Exp Hematol. 2007;35(7):1038–46. doi: 10.1016/j.exphem.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 45.Kim D, Cheng GZ, Lindsley CW, Yang H, Cheng JQ. Targeting the phosphatidylinositol–3 kinase/Akt pathway for the treatment of cancer. Curr Opin Investig Drugs. 2005;6(12):1250–1258. [PubMed] [Google Scholar]
- 46.Gills J, Holbeck S, Hollingshead M, et al. Spectrum of activity and molecular correlates of response to phosphatidylinositol ether lipid analogues, novel lipid-based inhibitors of Akt. Mol Cancer Ther. 2006;5(3):713–722. doi: 10.1158/1535-7163.MCT-05-0484. [DOI] [PubMed] [Google Scholar]
- 47.Castillo SS, Brognard J, Petukhov PA, et al. Preferential inhibition of Akt and killing of Akt-dependent cancer cells by rationally designed phophatidylinositol ether lipid analogues. Cancer res. 2004;64:2792–92. doi: 10.1158/0008-5472.can-03-1530. [DOI] [PubMed] [Google Scholar]
- 48.Calleja V, Alcor D, Laguerre M, et al. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007;5(4):e95. doi: 10.1371/journal.pbio.0050095. [DOI] [PMC free article] [PubMed] [Google Scholar]