




Abstract
Mapping genetic interactions (GIs) by simultaneously perturbing pairs of genes is a powerful tool for understanding complex biological phenomena. Here we describe an experimental platform for generating quantitative GI maps in mammalian cells using a combinatorial RNA interference strategy. We performed ~11,000 pairwise knockdowns in mouse fibroblasts, focusing on 130 factors involved in chromatin regulation to create a GI map. Comparison of the GI and protein-protein interaction (PPI) data revealed that pairs of genes exhibiting positive GIs and/or similar genetic profiles were predictive of the corresponding proteins being physically associated. The mammalian GI map identified pathways and complexes but also resolved functionally distinct submodules within larger protein complexes. By integrating GI and PPI data, we created a functional map of chromatin complexes in mouse fibroblasts, revealing that the PAF complex is a central player in the mammalian chromatin landscape.
Epistasis is a biological phenomenon in which the phenotype of one gene is modified by the presence or absence of another gene. Such relationships between genes, broadly termed GIs or epistatic interactions can be divided into three broad categories: negative (aggravating), whereby the resulting phenotype is more severe than is expected from the phenotypes associated with the single mutants; positive (alleviating), where the compound phenotype is less severe than anticipated; and neutral, where the measured phenotype is as expected1. A GI profile is a set of GIs for a given gene, and it reports on the functional relationships between cellular factors. Analyses of large numbers of profiles can reveal how groups of proteins and complexes work together to carry out higher-level biological functions1. Therefore, GIs have been very powerful in uncovering basic, mechanistic biology2,3 as well as in understanding the underlying causes of human disease4.
To date, the bulk of the available GI data has been generated in the yeast species, Saccharomyces cerevisiae and Schizosaccharomyces pombe5,6 and several experimental systems have been developed to generate GI maps in other model organisms, including Escherichia coli7, Caenorhabditis elegans8 and Drosophila melanogaster9. Large-scale GI data have been collected in S. cerevisiae using the synthetic genetic array approach initially qualitatively10 and later also quantitatively11. Building upon the synthetic genetic array approach, we developed epistatic miniarray profiling to quantitatively assess GIs. The resulting dataset using this approach, termed an epistatic mini-array profile (E-MAP), encompasses both positive and negative GIs in focused sets of genes, including those whose proteins are physically associated2 and/or function in the same process12. Quantitative GI mapping has revealed fundamentally important relationships between genes and has led to a better understanding of many biological processes. By analogy, applying it to mammalian cells will almost certainly reveal insight into human health and disease. We took advantage of recent developments in high-throughput RNA interference (RNAi) technology to develop an experimental platform for GI mapping in mammalian cells. We generated an E-MAP focused on 130 genes functioning in chromatin regulation in mouse fibroblast cells comprising ~11,000 quantitative GI measurements. By comparing GI data with known PPIs we found, as in simpler organisms, that GIs are strongly predictive of protein complexes and pathways. Furthermore, these integrated data allowed us to generate a functional connectivity map of mammalian protein complexes involved in chromatin regulation. Ultimately, this platform can be used to genetically interrogate functionally related sets of genes in a variety of mammalian cell types.
RESULTS
A pipeline for mapping of epistasis in mammalian cells
Our platform for the generation and quantification of GIs in mammalian cells (Fig. 1) is based on RNAi-mediated depletion of gene function in a pairwise fashion and measurement of the resulting phenotypic consequences. The availability of genome-wide RNAi libraries (small interfering RNA (siRNA), small hairpin RNA (shRNA) and endonuclease-prepared siRNA (esiRNA)) for higher organisms has enabled high-throughput genetic screens to be routinely performed in an arrayed or pooled setting13. Pooled approaches are based on monitoring the enrichment or depletion of specific sequence barcodes from a starting pool as a proxy for cell fitness14. In this study, we developed a platform in which the phenotypic effects of pairwise knockdowns can be quantified in a systematic manner. The measurement of every combination in the array is central in the subsequent data analysis.
Figure 1.

An overview of the mammalian E-MAP pipeline. Flowchart of the experimental setup: esiRNAs to a set of genes are arrayed in a pairwise fashion (in quadruplicate) in tissue culture plates. Reverse transfection is then performed, and the resulting fitness defects are observed using high-content imaging. Raw data are scored and phenotypic signatures are derived for each gene.
esiRNA are siRNAs generated by enzymatic cleavage of a long double-stranded RNA (dsRNA), resulting in a heterogeneous mixture of siRNA sequences that all target the same mRNA sequence15. This reagent has several properties that make it preferable over other approaches that rely on chemically synthesized siRNA and shRNA. Because any single siRNA sequence is present at an extremely low concentration, esiRNAs exhibit less off-target effects (and noise) than commonly associated with single siRNAs and shRNAs16. In addition, the number of pairs to test (and hence the amount of reagent required) grows quadratically with the number of genes to be interrogated, rendering the cost of siRNA or shRNA reagents prohibitive in an arrayed format. Conversely, large quantities of esiRNAs can be produced inexpensively in a high-throughput manner (Online Methods).
In our study, we used cell count as a measure of cell proliferation capacity or fitness. A conceptually similar readout (such as colony size) has been used in yeast and bacteria5,7,17. Data from these simpler systems suggests that strong GIs are rare10,12; therefore a general phenotype such as proliferation capacity, influenced by many different genes and pathways, is likely to uncover GIs. Finally, this phenotype is relatively easy to monitor with unsophisticated and affordable instrumentation.
To generate E-MAPs in mammalian cells, we dispensed unique esiRNAs into a 96-well or 384-well microplate (the array), and, using a custom-built liquid handler (Supplementary Fig. 1), a single constant esiRNA was overlaid (the query) to produce all pairwise combinations with the given query (Fig. 1). We transfected mouse fibroblast cells engineered to express a nuclear marker (histone macroH2A-EGFP), and allowed them to proliferate for 72 h. We assessed cellular proliferation by counting the number of cells containing fluorescent nuclei in each well using high-throughput fluorescence cytometry with an Acumen eX3 plate cytometer (TTP Labtech). We chose the microplate format over a cell spot microarray–based method18 as it allows many more cells to be monitored (thousands rather than dozens), thus allowing for the detection of more subtle phenotypes. We tailored protocols that allowed us to perform high-throughput knockdown experiments with a screening Z factor19 of 0.57 and >90% knockdown of EGFP as positive control (Supplementary Fig. 2a). As confirmation that GIs are indeed rare, addition of the query did not on average result in large deviations from the number of cells expected from single esiRNAs alone (Supplementary Fig. 2b), and we clearly observed fitness defects associated with particular queries when combined with the esiRNA array (Supplementary Fig. 2c).
We scored raw data using a GI score (termed S score) based on a neutral interaction model centered at zero (a neutral gene pair) and developed to capture a continuous spectrum of phenotype strengths20 (Fig. 1). We processed the data using a similar statistical framework similar to the one developed for yeast and bacterial GI mapping (Online Methods; http://sourceforge.net/projects/emap-toolbox/)17,20. A different GI scoring system, termed the π score, based on similar assumptions as the S score had been developed for analysis of RNAi-based GI data derived from D. melanogaster9. Using our mammalian data, we found that GIs obtained using the two methods were very similar (Supplementary Fig. 3a). However, based on our data, the S score was slightly more reproducible (Fig. 2a and Supplementary Fig. 3b), allowed for rational selection of score cutoffs based on a neutral interaction model and facilitated comparisons to other organisms for which similar data have been obtained, and therefore we used it to define our GIs.
Figure 2.
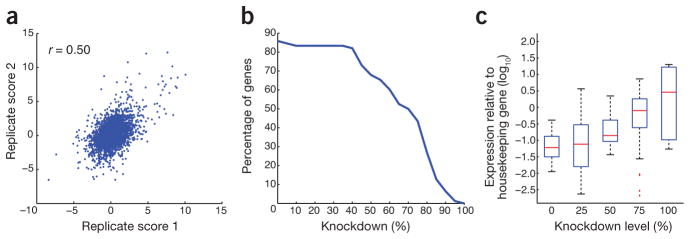
Characteristics and quality control of GI data set. (a) GI scores derived from independent biological replicate experiments. Each data point represents a GI score (S score) for the same pairwise knockdown derived from independent experiments. (b) Knockdown efficiencies as measured by quantitative reverse transcription PCR. (c) Relationship between knockdown efficiency and expression: data were split into five groups and plotted against gene expression relative to that of a housekeeping gene.
A chromatin-centered E-MAP in mouse fibroblasts
We used our epistasis-mapping pipeline to genetically interrogate factors involved in chromatin regulation in mice. We targeted 130 genes involved in chromatin or chromosome regulation using pairwise combinations of esiRNAs in mouse fibroblast cells (Online Methods). A common concern when using RNAi is that only partial phenotypes may be observed from incomplete knockdowns. However, hypomorphic (DAmP) alleles, which reduce protein levels of essential genes in yeast12,21, frequently yield rich and biologically meaningful GI profiles, suggesting that partial gene-product depletion produced by RNAi can be informative. We verified the efficiency of mRNA knockdown and determined that the transcripts for ~60% of the selected genes were inhibited by more than 60% (Fig. 2b and Supplementary Table 1). We found that knockdown efficiencies correlated with expression levels of the target in this cell line (Fig. 2c). Furthermore, we observed that efficient knockdown of individual genes occurred when two different esiRNAs were present (Supplementary Figs. 4 and 5). We generated a data set covering ~11,000 pairwise combinations (Supplementary Data 1 and 2) and the final data is of high quality as judged by internal consistency based on correlating S scores derived from independent biological replicates (Fig. 2a). Using an empirical cutoff for the S score, which reflects a high confidence of bona fide GIs20 (Supplementary Fig. 6), we uncovered 929 positive (S ≥ 2) and 611 negative (S ≤ −2) GIs (Supplementary Table 2). Finally, to assess potential off-target effects and help address the overall quality of the data set, we generated independent esiRNAs for five of the genes on the E-MAP (Cnot1, Cnot2, Cnot3, Paf1 and Leo1; Supplementary Table 3) and found that the genetic profiles they generated were highly correlated with the profiles from the original esiRNA constructs (ranging from r = 0.61 to r = 0.84; Supplementary Fig. 7). The data from these experiments were averaged and integrated into the final data set (Supplementary Data 2).
Comparison of mammalian GI and PPI data
Epistasis mapping is a powerful tool for unbiased discovery of protein complexes, larger functional modules and pathways. Data from simpler systems, including S. cerevisiae and S. pombe, suggest that genes in functional modules exhibit two important characteristics: (i) their phenotypic signature or profile of GIs tend to be highly correlated; and (ii) the corresponding genes are enriched for positive (suppressive) GIs1. We observed similar trends in the mammalian data set using a set of known protein complexes and pathways (Supplementary Table 4). Indeed, pairs of genes whose products function together or are in the same complex were likely to interact positively (Fig. 3a) and have highly correlated phenotypic signatures (Fig. 3b). We also found that GI scores and profiles were strongly predictive of pairs of genes that are involved in the same functional module using an unbiased set of interactions based on probabilistic functional gene networks (HumanNet) at a range of cutoff values22 (Supplementary Fig. 8).
Figure 3.
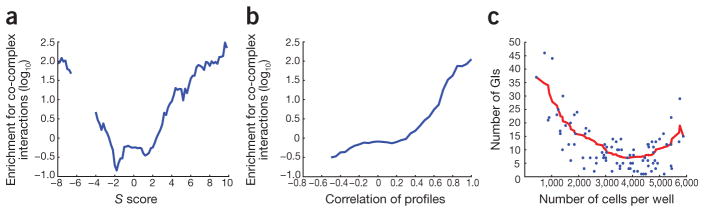
Comparison of GI and PPI data sets. (a,b) Comparison of individual S scores (a) and GI profiles (b) to the likelihood of the corresponding pairs of proteins being physically associated. (c) Fitness after single gene knockdown versus the number of genetic interactions associated with the same gene. Raw cell counts were used as a proxy for fitness, and a cutoff of S ≥ 2 and S ≤ −2 was used to define informative GIs on the y axis.
We observed enrichment for strongly negative GIs and PPIs between pairs of factors (Fig. 3a). These trends could be due to the fact that RNAi often only partially disables the function of a given protein. In these cases, an additional perturbation of another protein in the same module could result in complete abolishment of its function, and therefore a negative GI would be observed. Consistent with this reasoning is the observation that GIs in yeast derived from hypomorphic alleles of essential genes were more likely to produce negative GIs when the corresponding proteins are physically associated3,11,23. Alternatively, these genetic relationships could correspond to non-essential genes whose proteins function in essential complexes. Additional data will need to be collected on a more global scale to study these relationships between PPI and GIs in more complex organisms. Finally, we observed a strong relationship between the fitness defect associated with a single gene knockdown and the number of GIs associated with that gene (Fig. 3c)5,24,25, suggesting that genes with functional interactions with many other genes have the most dramatic effect on proliferative phenotypes. This is an important observation as it suggests that initial single RNAi screens could enable the design of more cost-effective, signal-rich E-MAPs.
A chromatin module map derived from the E-MAP
GI data from the E-MAP allow for the identification of individual functional modules at the level of individual S scores or by hierarchical clustering of GI profiles. For example, the five members of the PAF complex (encoded by Paf1, Leo1, Ctr9, Cdc73 and Rtf1), involved in transcriptional elongation and chromatin modification26,27, display similar GI profiles and positive GIs with each other (Fig. 4a). In addition, Supt16h, a component of the FACT complex, a chromatin-specific elongation factor28, was also in this functional module, consistent with the previous findings that the PAF complex is physically and functionally connected to FACT in budding yeast27.
Figure 4.

A module map of chromatin-related genes. (a) GIs (left) and profile Pearson correlations (right) for members of the PAF transcriptional elongation complex. (b) GIs (left) and profile Pearson correlations (right) for members of the CNOT complex. S scores and profile Pearson correlations were used in a and b. (c) A module map based on a manually curated set of protein complexes (Supplementary Table 4). Modules and inter-module GI bundles are colored according to the enrichment of the observed GIs, with gray signifying no enrichment of a particular interaction type was observed (Supplementary Table 5). (d) Examples of positive and negative GI bundles corresponding to particular edges on the module map.
Our method also can be used to resolve functionally distinct modules in complexes. For example, members of the CNOT complex, involved in mRNA deadenylation and chromatin modification29, form two distinct groups: a highly correlated, positively interacting module comprising products of Cnot1, Cnot2 and Cnot3 (CNOT core) and an uncorrelated group containing products of Cnot4, Cnot6, Cnot7 and Cnot8 (Fig. 4b). Consistent with this observation, a recent report demonstrated that Cnot1, Cnot2 and Cnot3 gene products behave as a functionally and physically distinct submodule within CNOT complex and the other components, including products of Cnot4, Cnot6 and Cnot8, bind to it in a dynamically regulated fashion30. These results confirm that mammalian GI maps have the ability to dissect larger, functionally disparate complexes, as has been observed in simpler eukaryotic organisms2.
Based on our manually curated set of protein complexes and pathways, we created a map depicting the genetic cross-talk between different functional modules represented in the E-MAP (Fig. 4c and Supplementary Tables 4 and 5). Contained within this map are modules corresponding to the INO80 chromatin remodeling complex31; the transcriptional regulator Integrator32; the MCM DNA replication complex; and cohesin and condensin33–35 complexes, involved in chromatin segregation and condensation. Notably, the PAF complex occupies a central position on the map, interacting strongly, both positively and negatively, with many other functional modules. For example, we observed strong positive GIs between the PAF complex and modules corresponding to CNOT and the RUVBL ATPase subcomplex (encoded by Ruvbl1, Ruvbl2 and Actl6a), which is part of several chromatin-remodeling complexes36 (Fig. 4d). Furthermore, we observed negative interactions between components of the PAF complex and the PARP (encoded by Parp1 and Parp2) and condensin (encoded by Smc2 and Smc4) modules (Fig. 4d). To explore the genetic relationship between the PAF complex and PARP modules, we treated cells that are depleted for PAF complex function (using RNAi-mediated knockdown of Rtf1) with the PARP inhibitor, Veliparib (ABT-888)37 (Supplementary Fig. 9). We observed a dose-dependent inhibition of growth specifically in the context of Rtf1 knockdown, consistent with the negative GIs we observed between PAF complex members and PARP (Supplementary Fig. 9). These data demonstrate that synthetic epistatic interactions can be leveraged to identify parallel pathways, which can be targeted using small molecules.
Additional investigation of the molecular basis of these individual connections will improve our understanding of how interactions among chromatin machines control the program of gene expression in mammals.
Validation of GIs using an orthogonal phenotypic readout
Phenotypic readouts that use growth rate to enumerate the proliferative capacity of a cell have been used in many GI screens5,7,17. However, E-MAPs can potentially be constructed using any phenotype that can be observed by microscopy, including subtle effects on cell morphology or structure. Because the impact of one gene on another may differ depending on which pathways influence the phenotype being measured, E-MAPs generated using different readouts may not necessarily be similar to each other but are highly complementary in understanding gene function38. However, we would expect partial overlap with our proliferation-based E-MAP and other phenotypic readouts because many different genes and pathways impinge on cells’ proliferation capacities. As the PAF complex is highly connected in the module map (Fig. 4c), we asked whether its epistatic interactions with other modules might be maintained when using alternate readouts. Using high-content microscopic interrogation, we observed that depletion of individual PAF-complex components leads to a very specific morphology phenotype in which cells display elongated nuclei (Fig. 5). This nuclear elongation may be due to disrupted regulation of the chromatin modification landscape as the PAF complex is required for normal histone H3 methylation at lysines 4, 36 and 79 (ref. 39).
Figure 5.

Validation of observed GIs with an orthogonal phenotypic readout. (a) Micrographs derived from mouse embryo fibroblast cells depleted for Ctr9 in combination with knockdowns of Ruvbl1, Ruvbl2, Actl6a or Morfl1. Scale bars, 20 μm. (b) Phenotype strength represented as a fraction of cells with elongated nuclei (longer bars represent more extreme phenotype). Error bars, s.d. based on 100 random samples of 10% of the data in each experiment.
We next tested the positive GIs that we had observed using the fitness-based readout between PAF complex components and the RUVB module. Combining a Ctr9 knockdown with knockdown of Ruvbl1, Ruvbl2 or Actl6a suppressed the elongated-nuclei phenotype (Fig. 5a,b), consistent with the positive interactions observed in our E-MAP (Fig. 4c,d). We did not see suppression of this specific phenotype when knockdown of Ctr9 was combined with knockdowns of other non-interacting factors, including Morf4l1 (Fig. 5 and data not shown), arguing there is a unique interaction between the PAF complex and the RUVBL module components. Although additional work will be required to understand why depletion of the PAF complex results in elongated nuclei and how absence of the RUVBL module suppresses this effect, these data demonstrate that we could recapitulate GIs with an orthogonal phenotypic readout.
DISCUSSION
Development of high-throughput epistasis-mapping technologies has made it possible to interrogate complex biological phenomena. Although methodologies for mapping PPIs are currently more widely used, these networks are still limited in that they only report on gene products that interact physically. GIs, in contrast, illuminate functional relationships between genes including, but not limited to, physical interactions of their gene products. They often reveal how groups of proteins and complexes work together to carry out biological functions and can describe the cross-talk between pathways and processes1. Therefore, GI networks are a natural complement to PPI maps and integrating these two types of information has proven to be extremely powerful in understanding complex biology in a variety of systems2,3,23.
Our platform, together with similar strategies developed for flies and worms8,9, is a great addition to the tools available to study higher eukaryotes, one that is easily adaptable to different mammalian cell types. By combining this platform with recent developments in high-content microscopy, one can exploit multidimensional (multifeature) phenotypes40, which will undoubtedly be of great utility in detailed dissection of various processes in mammalian cells. Ultimately, these data sets can be generated under different conditions to create differential E-MAPs24 to widen the GI space and deepen the interrogation of specific pathways and processes. Our platform can also be used to characterize aberrant pathways associated with different disease states, such as cancer, using specific, mutated cell lines. Finally, our approach can be exploited to generate GI data sets to interrogate sets of genes known to be targeted by a pathogen during infection. The E-MAPs can then be generated in the presence or absence of the pathogenic organism (where different phenotypic characteristics of both the pathogen and host can be monitored) in an effort to gain a deeper understanding of how the host machinery is hijacked and rewired during the course of infection.
ONLINE METHODS
Tissue culture
Standard media formulations were used throughout. Experiments were performed in 96- or 384-well tissue-culture plates (Greiner).
Cell lines
A mouse fibroblast cell line stably expressing nuclear-localized histone macroH2A-EGFP construct was used41,42.
cDNA preparation
cDNA used for esiRNA preparation was prepared from mouse cells from RNA purified using Trizol (Invitrogen) and standard protocols. First-strand synthesis was primed with oligo(dT).
esiRNA preparation
esiRNA was prepared essentially as described previously15. Oligo sequences are listed in Supplementary Table 3. Below is a detailed description as this is a multistep process and some of the steps were optimized for 96-well format preparations.
First-round PCR
We used first-strand cDNA from target cells as a template for first-round PCR (35 cycles). Primers for first round must begin with T7 ‘anchor’ sequence: 5′-GGGCGGGT-3′ to which the T7 Anchor primer will anneal in the second round. One microliter of 1/20 dilution of first PCR per 50 μl reaction is used as template for second-round PCR to incorporate T7 promoters. Cycling conditions were as follows: 94 °C, 2 min, 35 × (94 °C, for 30 s, 55 °C for 45 s, 72 °C for 1 min), 72 °C for 5 min, hold at 4 °C.
Second-round PCR
A pair of T7 anchor primers (5′-TAATACGACTCACTATAGGGAGACCACGGGCGGGT-3′) was used to attach T7 promoter sequences used in the subsequent in vitro transcription (IVT). Cycling conditions were as follows: 94 °C for 2 min, 5 × (94 °C for 30 s, 42 °C for 45 s, 72 °C for 1 min), 30 × (94 °C for 30 s, 60 °C for 45 s, 72 °C for 1 min), 72 °C for 5 min, hold at 4 °C.
In vitro transcription and annealing
In IVT reactions, we used the second-round PCR product as template. Annealing was done using the same program immediately after the IVT reactions. Transcription-annealing program was as follows: 37 °C for 5.5 h, 90 °C for 3 min, ramp (0.1 °C s−1) to 70 °C, 70 °C for 3 min, ramp (0.1 °C s−1) to 50 °C, 50 °C for 3 min, ramp (0.1 °C s−1) to 25 °C, 25 °C for 3 min, hold at 4 °C. Products were frozen at −20 °C or were purified on Qiagen RNeasy column (or Invitrogen PureLink Micro-to-Midi purification column). The dsRNA products were relatively small and may not be quantitatively precipitated with ‘standard’ purification procedures. If products are < 250 base pairs, ethanol concentration for precipitation was increased to 50%, and we advise skipping any washes with low ethanol (that is, wash buffer 1). Products were eluted off of columns with 2 or 3 serial elutions, and we extended elution times of 5 min or more per elution. Concentration of esiRNAs was quantified with Nanodrop. We ran 1 μl of the products on a 1% agarose gel to determine whether the band appeared as a single band rather than as a smear. Yield is directly proportional to the amount of template, that is, poor yield of secondary PCR results in bad IVT yield. We confirmed robust second-round PCR before IVT.
RNase III digestion and esiRNA purification
Purified dsRNA was digested in 100-μl reactions as below using NEB ShortCut RNase III (use ~1.3 U – 1.6 U per μg purified dsRNA). Digestion conditions were 37 °C for 30 min, then reactions were placed on ice, 1/10th volume 0.5 M EDTA was added. Purification was done immediately after RNase III digestion over two columns (first to remove undigested dsRNA and secondary PCR product (33% ethanol final), second to concentrate esiRNA (75% ethanol final)). Either RNeasy (Qiagen) columns or PureLink Micro-to-Midi (Invitrogen) purification columns can be used. As this is a critical step, a detailed purification protocol assuming a 100 μl RNase III digest is given below. Wash volumes are given per 96-well plate. We added 110 μl lysis buffer and mixed the reactions; then we added 110 μl 100% ethanol (33% ethanol final), mixed and loaded the reaction onto a first mini column. Next we centrifuged the reaction for 30 s at 8,000 r.p.m. in a standard Eppendorf benchtop centrifuge, collected flow-through, added 583 μl 100% ethanol to flow-through, mixed (final ethanol was 75%) and loaded the reaction onto a second column, which we centrifuged 30 s at 8,000 r.p.m. Next we washed the column with 750 μl wash buffer II or RPE (to have 80% ethanol final), centrifuged for 1 min at 8,000 r.pm., washed column with 500 μl wash buffer II or RPE and centrifuged spin column at maximum speed for 2 min. The column was transferred to a collection tube and air-dried ~2 min. Column was eluted with 3 × 50 μl H2O with extended elution time (>15 min), and product was quantified with Nanodrop. We ran 1 μl of the product on a nondenaturing 15% acrylamide gel (0.5 or 1× TBE). A distribution of the product between two bands was usually seen: about two-thirds of the RNA within a band of ~25–27 bp and about one-third within a 12–15 bp band. This lighter band did not elicit knockdown but did not interfere with knockdown either. Often a small smearing up to about 30 bp was observed, but it was usually minimal. The digestion was dependent on dsRNA:enzyme ratio and time. Extended digestion reduced yield considerably. Typical yield was 50–70% of input.
Array preparation
We dispensed 7.5 ng of an array of 96 esiRNAs at 0.75 ng/μl in 384-well tissue-culture plates in quadruplicate using a Biomek FX liquid handler. Then, 7.5 ng of the query esiRNA was dispensed into each well using a Multidrop Nano (Thermo) liquid dispenser attached to a custom-built liquid handler (Supplementary Fig. 1). The plates were vacuum-dried and could be stored without apparent loss of activity until transfected.
Transfection of X3 macroH2A-EGFP cells
Lipid-based transfection using Lipofectamine RNAi MAX (Invitrogen) was used. For 384-well format tissue-culture plates 750–1,000 cells/well were transfected with 15 ng (7.5 ng each) esiRNA using 0.1 μl transfection reagent per well. Lipid-RNA complexes were reconstituted in OptiMEM for 15–30 min before cell addition. Cells were added in antibiotic-free DMEM supplemented with 10% FBS and incubated overnight. Before we sealed the plates, fresh DMEM supplemented with penicillin, streptomycin and 10% FBS was added.
Quantitative reverse transcription–PCR
cDNA was prepared using CellSure cDNA kit (Bioline) per manufacturer’s instructions. Quantitative reverse transcription (RT)-PCR was carried out using SensiFAST SYBR lo-ROX master mix (Bioline) on a MX4000 qPCR instrument (Stratagene).
Raw-data collection
Cell counting was done using an Acumen eX3 plate cytometer (TTP Labtech) using 384-well tissue culture plates. Data were collected in batches of more than 20 screens each.
Scoring of GIs
Raw data were scored using a published software toolbox20. Individual batches were normalized and scored separately, thus minimizing systematic experimental biases and batch-to-batch variation.
Detailed explanation of the S score
Normalization of cell numbers
Raw cell numbers were normalized to correct for differences in growth conditions. The normalizations used here were multiplicative normalizations. We tried other normalization methods as well (including a logarithmic normalization) and found them to be less effective. For each plate, a value referred to as the plate middle mean (PMM) was computed as the mean of the cell numbers ranked in the 40th to 60th percentile of the cell number on the plate, excluding the outermost two rows and columns on the plate. The number of cells in the outermost two rows and columns were then scaled such that the median number of cells of each such row or column was equal to the PMM. Cell numbers in the outermost rows and columns were treated as a special case because the data there tended to be noisier than in the center of the plate, and this extra variation tended to be consistent across each such row and column (that is, the entire top row might uniformly be unusually large or small).
Batch-to-batch variability has been noted in previous GI screens, where the typical cell number estimated for one group of screens completed at approximately the same time using the same set of reagents differ from the values estimated for another group of screens completed at a different time (perhaps weeks or months apart). To remove this batch variability, we processed each set of screens done on the same date (>20 screens) as a subset as we have done previously17. When the data were processed and analyzed without taking into account batch correction, the internal correlation of independent biological replicates was reduced from 0.50 to 0.30 (data not shown). The S scores from each individual subset were combined to create the larger, final data set. In the case of duplicate S scores (that is, from duplicate screens in different batches), S scores were averaged.
A natural question is then how many different screens need to be included in a batch, such that the estimated expected values will be reliable? Experience from previous E-MAPs in yeast suggests that as increasingly many screens are completed in the same batch, the error in estimates of expected cell numbers decreases (data not shown). There can be substantial error if a batch includes fewer than 20 screens. On the other hand, each additional screen beyond about the 40th gives only marginal improvement (data not shown).
Scoring GIs
Double knockdowns were scored as to the magnitude and sign of the observed GI. We wanted a score that would reflect both our confidence in the presence of GIs as well as the strengths of interactions, and so we chose to use a modified t-value score (S). A standard t value is computed as:
where
where μExp is the mean of normalized cell numbers for the double knockdown of interest; varExp is the variance of the normalized cell number for the double knockdown of interest; nExp is number of measurements; μCont is the mean of normalized cell numbers for the control array single knockdown corresponding to the double knockdown of interest; varCont is the variance of the normalized cell numbers for this control knockdown; and nCont is the number of measurements for the control knockdown. The S score is constructed in the same way:
where
but with the following modifications: μCont is median of normalized cell numbers for all double knockdowns containing a particular esiRNA; varExp is the maximum of the variance of normalized cell number for the double knockdown of interest or a minimum bound described below; varCont is the median of the variances in normalized cell numbers observed for all double knockdowns containing the esiRNA of interest or a minimum bound described below; and nCont = 4 (this was the median number of experimental replicates over all the experiments).
Minimum bound on varExp
A minimum bound was placed on the experimental s.d. (and hence on the variance) because we observed that occasionally, by chance, repeated measurements would give an unusually small s.d., resulting in a large score, but these large scores did not seem to be reproducible, nor did they reflect strong GIs. We therefore placed a minimum bound on this s.d. equal to the expected s.d. in normalized cell numbers for a double knockdown derived from esiRNA with similar growth phenotypes. The expected s.d. was calculated by measuring the observed standard errors in measurement as a function of cell numbers typical for one or the other esiRNA knockdown.
Minimum bound on varCont
For similar reasons as for varExp and because it improved the reproducibility of computed S scores, a lower bound was also placed on varCont. This lower bound was equal to μCont multiplied by the observed median relative error (standard deviation divided by mean cell number) for all measurements in the data set.
Note concerning normalization using the PMM and scoring using the median values
Both of these measures may be biased if the frequency of synthetic interactions is significantly greater or smaller than the frequency of alleviating interactions for a particular gene. However, we have observed this bias to be relatively small (data not shown), and we include in our Matlab toolbox an alternative strategy to estimate the typical cell number on an experimental plate or for a given esiRNA knockdown. The alternative strategy, which uses a Parzen window approach to estimate the most common cell number, is less sensitive to skewed distributions of cell numbers.
Data processing using E-MAP toolbox
To launch the graphical user interface, “emapGUI” was typed at the command prompt in Matlab. The software offers several different preprocessing and normalization options. We used the options menu to set those. The following were used for processing the raw data: normalization method, Parzen; control size method: Parzen; preprocessing option: no preprocessing; threshold for removing frequently missing data: 0.15; filter data with missing neighbors: no filter. We used the ‘File’ --> ‘load raw data’ command to load data from source files (see the included example files for templates for making the necessary coordinate and file name maps). We used ‘Data processing … score data’ to generate unaveraged S scores. We used ‘File’ --> ‘Save as MAT file’ to save the analysis. Individual batches were scored separately, and the resulting score tables (scoremat structure in the .mat file) were then merged to create the final score matrix. Data for replicate screens that correlated at ≤ 0.1 were removed from the dataset.
HCS microscopy
A Cell Insight Personal Image Cytometer (Thermo) with a 10× objective was used to collect microscopy data. Image analysis was performed using the Cellomics vHCS Discovery Toolbox (Thermo). Nuclear shape in Figure 5 was defined as a length to width ratio for each cell.
Module map
Curated protein complexes and pathways were integrated withS scores from this study. Between-module interactions were assessed by comparison of the average S score of GIs between two modules to 10,000 sets of random interactions of the same size as in ref. 23. Displayed edges had P < 0.01.
Supplementary Material
Acknowledgments
We thank members of the Bandyopadhyay, Panning and Krogan laboratories for constructive discussions, J. Forrest (TTP Labtech) for providing the Acumen eX3 cytometer, J. DeRisi for allowing us access to the uPrint 3D printer, G. Tzertzinis (NEB) for providing high-concentration ShortCut RNase III, and T. Fazzio for technical advice. This work has been supported by US National Institutes of Health (GM082250, GM081879, AI090935, AI091575 to N.J.K., P50CA58207 to S.B. and GM085186 to B.P.); Defense Advanced Research Projects Agency (HR0011-11-C-0094 to N.J.K.) and University of California San Francisco Program for Breakthrough Biomedical Research to N.J.K. and B.P.
Footnotes
Supplementary information is available in the online version of the paper.
AUTHOR CONTRIBUTIONS
A.R., D.T., G.C., S.B., B.P. and N.J.K. designed the project; A.R. and D.T. performed the experiments; A.R., D.T., M.S., G.L.N. and S.B. analyzed the data; and A.R., D.T., M.S., G.C., S.B., B.P. and N.J.K. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Beltrao P, Cagney G, Krogan NJ. Quantitative genetic interactions reveal biological modularity. Cell. 2010;141:739–745. doi: 10.1016/j.cell.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins SR, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–810. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 3.Wilmes GM, et al. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell. 2008;32:735–746. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehner B. Modelling genotype-phenotype relationships and human disease with genetic interaction networks. J Exp Biol. 2007;210:1559–1566. doi: 10.1242/jeb.002311. [DOI] [PubMed] [Google Scholar]
- 5.Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan CJ, et al. Hierarchical modularity and the evolution of genetic interactomes across species. Mol Cell. 2012;46:691–704. doi: 10.1016/j.molcel.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Typas A, et al. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 9.Horn T, et al. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat Methods. 2011;8:341–346. doi: 10.1038/nmeth.1581. [DOI] [PubMed] [Google Scholar]
- 10.Tong AH, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 11.Baryshnikova A, et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods. 2010;7:1017–1024. doi: 10.1038/nmeth.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuldiner M, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 13.Mohr S, Bakal C, Perrimon N. Genomic screening with RNAi: results and challenges. Annu Rev Biochem. 2010;79:37–64. doi: 10.1146/annurev-biochem-060408-092949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sims D, et al. High-throughput RNA interference screening using pooled shRNA libraries and next generation sequencing. Genome Biol. 2011;12:R104. doi: 10.1186/gb-2011-12-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buchholz F, Kittler R, Slabicki M, Theis M. Enzymatically prepared RNAi libraries. Nat Methods. 2006;3:696–700. doi: 10.1038/nmeth912. [DOI] [PubMed] [Google Scholar]
- 16.Kittler R, et al. Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nat Methods. 2007;4:337–344. doi: 10.1038/nmeth1025. [DOI] [PubMed] [Google Scholar]
- 17.Collins SR, Roguev A, Krogan NJ. Quantitative genetic interaction mapping using the E-MAP approach. Methods Enzymol. 2010;470:205–231. doi: 10.1016/S0076-6879(10)70009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rantala JK, et al. A cell spot microarray method for production of high density siRNA transfection microarrays. BMC Genomics. 2011;12:162. doi: 10.1186/1471-2164-12-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 20.Collins SR, Schuldiner M, Krogan NJ, Weissman JS. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 2006;7:R63. doi: 10.1186/gb-2006-7-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breslow DK, et al. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee I, Blom UM, Wang PI, Shim JE, Marcotte EM. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011;21:1109–1121. doi: 10.1101/gr.118992.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bandyopadhyay S, Kelley R, Krogan NJ, Ideker T. Functional maps of protein complexes from quantitative genetic interaction data. PLOS Comput Biol. 2008;4:e1000065. doi: 10.1371/journal.pcbi.1000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandyopadhyay S, et al. Rewiring of genetic networks in response to DNA damage. Science. 2010;330:1385–1389. doi: 10.1126/science.1195618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch EN, et al. Conserved rules govern genetic interaction degree across species. Genome Biol. 2012;13:R57. doi: 10.1186/gb-2012-13-7-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding L, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell. 2009;4:403–415. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Krogan NJ, et al. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol. 2002;22:6979–6992. doi: 10.1128/MCB.22.20.6979-6992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belotserkovskaya R, Reinberg D. Facts about FACT and transcript elongation through chromatin. Curr Opin Genet Dev. 2004;14:139–146. doi: 10.1016/j.gde.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Collart MA, Panasenko OO. The Ccr4–not complex. Gene. 2012;492:42–53. doi: 10.1016/j.gene.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 30.Lau NC, et al. Human Ccr4-Not complexes contain variable deadenylase subunits. Biochem J. 2009;422:443–453. doi: 10.1042/BJ20090500. [DOI] [PubMed] [Google Scholar]
- 31.Conaway RC, Conaway JW. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci. 2009;34:71–77. doi: 10.1016/j.tibs.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Wagner EJ. snRNA 3′ end formation: the dawn of the Integrator complex. Biochem Soc Trans. 2010;38:1082–1087. doi: 10.1042/BST0381082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol. 2005;6:476–486. doi: 10.1038/nrm1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 35.Cuylen S, Haering CH. Deciphering condensin action during chromosome segregation. Trends Cell Biol. 2011;21:552–559. doi: 10.1016/j.tcb.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 36.Gallant P. Control of transcription by Pontin and Reptin. Trends Cell Biol. 2007;17:187–192. doi: 10.1016/j.tcb.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Murai J, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michaut M, Bader GD. Multiple genetic interaction experiments provide complementary information useful for gene function prediction. PLOS Comput Biol. 2012;8:e1002559. doi: 10.1371/journal.pcbi.1002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta. 2010;1799:379–388. doi: 10.1016/j.bbagrm.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loo LH, Wu LF, Altschuler SJ. Image-based multivariate profiling of drug responses from single cells. Nat Methods. 2007;4:445–453. doi: 10.1038/nmeth1032. [DOI] [PubMed] [Google Scholar]
- 41.Marciniak RA, Lombard DB, Johnson FB, Guarente L. Nucleolar localization of the Werner syndrome protein in human cells. Proc Natl Acad Sci USA. 1998;95:6887–6892. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nusinow DA, et al. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, MacroH2A, and contributes to silencing of the inactive X chromosome. J Biol Chem. 2007;282:12851–12859. doi: 10.1074/jbc.M610502200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.