




Abstract
The Matchmaker Exchange API allows searching a patient's genotypic or phenotypic profiles across clinical sites, for the purposes of cohort discovery and variant-disease causal validation. This API can be used not only to search for matching patients, but also to match against public disease and model organism data. This public disease data enables matching known diseases and variant-phenotype associations using phenotype semantic similarity algorithms developed by the Monarch Initiative. The model data can provide additional evidence to aid diagnosis, suggest relevant models for disease mechanism and treatment exploration, and identify collaborators across the translational divide. The Monarch Initiative provides an implementation of this API for searching multiple integrated sources of data that contextualize the knowledge about any given patient or patient family into the greater biomedical knowledge landscape. While this corpus of data can aid diagnosis, it is also the beginning of research to improve understanding of rare human diseases.
Keywords: rare disease, informatics, ontology, phenotype, model systems, Matchmaker Exchange
Introduction
The past two decades have witnessed enormous progress in our understanding of the genome due to large-scale projects that have interrogated the sequence and variation of the human genome as well as functional genomics projects. The need to understand the relationships between genomic variation and human disease has brought about a number of large-scale projects such as UK 100,000 Genomes (http://www.genomicsengland.co.uk/the-100000-genomes-project/), NIH Undiagnosed Diseases Program/Network (Tifft and Adams 2014), The Cancer Genome Atlas (Weinstein et al. 2013), and others. However, despite such efforts, we know the phenotypic consequences of only approximately 38% of the human coding genome, and associated genes have not been identified for approximately half of known heritable diseases (Boycott et al. 2013). Further, since each of us harbor some 100 genuine loss of function variants with around 20 genes completely inactivated (MacArthur et al. 2012), prioritization based solely on variant frequency and pathogenicity cannot reliably identify the causative mutation in all cases – the ability to compute on phenotype as well as sequence is necessary.
Phenotyping in humans and model organisms
There are an ever-increasing number of large-scale projects to catalog phenotypic abnormalities in model organisms, for example the International Mouse Phenotyping Consortium (IMPC) (Koscielny et al. 2014) and comparable efforts in Zebrafish (Kettleborough et al. 2013). This is particularly relevant for rare and undiagnosed diseases, since phenotype data is available from rat, mouse, zebrafish, worm, and fruitfly via orthology for approximately 80% of the human coding portion of the genome. However, similar to human informatics resources, much of the genomic data in such projects is standardized for exchange and computation but the phenotypic data remains fairly unstructured, is very diverse, and is much less amenable to computation.
The development of resources for the computational analysis of disease has been substantially slower for a number of reasons, mainly including the complexity of computational representations of disease manifestations (phenotype), of disease causation (etiology), and of the development of disease manifestations and complications with time (disease course and natural history). Deep phenotyping (the precise and comprehensive analysis of phenotypic abnormalities in which the individual components of the phenotype are observed and described), represents an important prerequisite for the success of the precision medicine endeavor. To maximize the utility of the results, deep phenotyping requires three main components: (i) controlled vocabularies or ontologies to precisely, accurately, and comprehensively describe phenotypic abnormalities in humans and model organisms (see Figure 1); (ii) use of these controlled vocabularies to describe disease manifestations and thereby provide computational models of human diseases or of medically relevant animal models of disease; and (iii) algorithms and tools that represent a foundation for the computational analysis of disease.
Figure 1. Example of a patient annotation to a subset of the Human Phenotype Ontology (HPO).
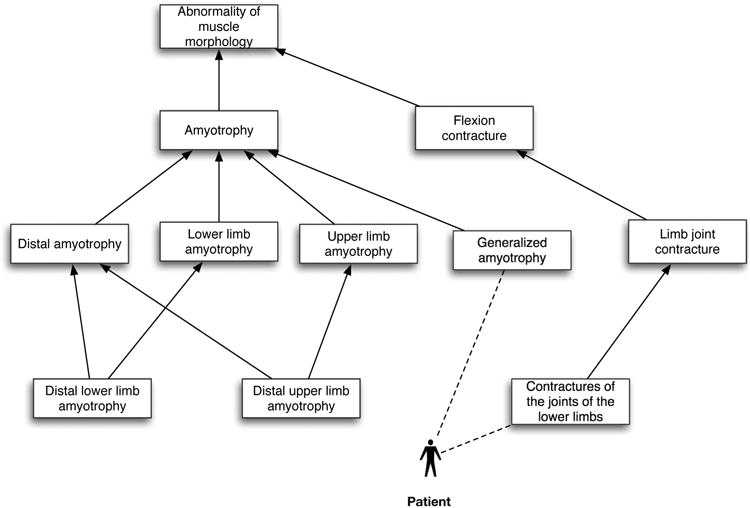
A hypothetical patient is annotated with two phenotypes, ‘Generalized amyotrophy’ and ‘Contractures of the joints of the lower limbs’ (annotations are indicated using dashed lines). Phenotypes can be described at different levels of granularity or specificity (more general terms are shown near the top of the figure). Any individual patient can be assigned any number of HPO terms.
The Monarch Initiative is an international consortium that aims to utilize improved deep phenotyping for the purposes of disease diagnostics and mechanism discovery. We provide vocabularies for the description of patients that are integrated with model organism vocabularies, and a system for computing over these descriptions. These vocabularies are intended to meet the demonstrated need for deep phenotyping vocabularies that focus on scientific investigation of the patient rather than billing or quality of care estimates (for example, the International Classification of Diesase ICD). In support of this goal, the Human Phenotype Ontology (HPO)(Robinson et al. 2008; Köhler et al. 2014) not only provides more granular patient phenotypes, but also because of its underlying logic, can be utilized for phenotypic comparison of model systems. We have similarly helped develop the Mammalian Phenotype Ontology (Smith and Eppig 2015), the zebrafish ontology (Van Slyke et al. 2014) and many others that have been semantically integrated (Mungall et al. 2010) with HPO to assist in the use of deep phenotyping data from model organisms in combination with human data. Towards this end, we have also constructed a large data corpus (www.monarchinitiative.org) of genotype-phenotype associations from human clinical sources and model organism sources, which have been semantically integrated using the aforementioned phenotype ontologies, together with a suite of genotype, anatomy, and sequence ontologies. The complete set of sources (currently 18) integrated is visible on http://monarchinitiative.org/sources. This corpus is the basis for the semantic similarity algorithms that have been implemented in tools such as Exomiser (Robinson et al. 2013) and PhenIX (Zemojtel et al. 2014) for the purposes of clinical variant prioritization. In these tools, clinical exome sequencing produces a list of candidates that can be further prioritized by utilizing what is known about the phenotypic effects of orthologous genes and interacting proteins in other species. These tools make use of the graph nature of the phenotype ontologies in order to score a match of an overall set of phenotypes (Robinson and Webber 2014).
Matchmaker Exchange
The Matchmaker Exchange (MME) API (Application Programming Interface) allows for the discovery of patients with shared genetic or phenotypic profiles across different sites (Buske et al., 2015; Phillipakis et al., 2015). Although it is possible for a Matchmaker to implement only gene matching (for example, Sobreira et al., 2015), we focus here on phenotype matching. The phenotypic profile of a patient (i.e. the set of individual phenotypes exhibited throughout the course of their disease or disorder) and/or their genotype at one site is matched against profiles at the partner site. The closest matching patients are returned, together with information about which portion of the profile matches. Sites can be paired in a variety of configurations, with a key exchange mechanism and tiered levels of availability of genotype and phenotype data to support privacy.
While the API has been designed and constructed with the specific purpose of performing patient-to-patient matchmaking, in practice, it can be extended to other use cases, such as patient-to-disease and patient-to-model matchmaking. This is because, conceptually, the elements being matched all have phenotype profiles, independently of the encapsulating entity - i.e., patient data, disease description or model system. Figure 2 shows the placement and interaction of the Monarch approach within a hypothetical constellation of Matchmakers. Here we review the Monarch matchmaking service and the implementation of the MME API.
Figure 2. Monarch in the MME Landscape.
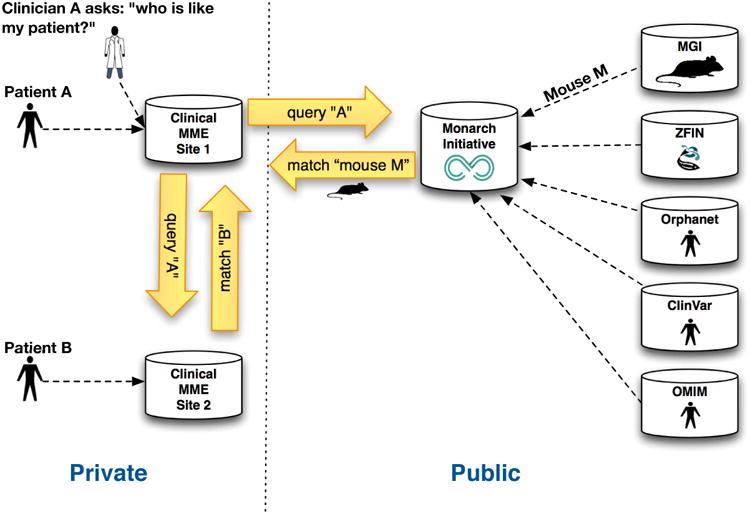
The MME API is used to facilitate discovery of patient B in a remote patient MME database, for a particular patient A that exhibits matching phenotypic features. The same clinical site can connect to Monarch to discover a range of models (for example, mouse and zebrafish) and other aggregated diseases and variants associated with similar phenotype profiles. Note the diagram only shows a subset of the many phenotypic knowledge sources feeding into the Monarch platform.
Results
The Monarch Matchmaking System
The Monarch disease-model comparison system is based on a large curated and aggregated knowledge base of phenotypic effects of variants covering both humans and model organisms such as mouse, zebrafish and drosophila. These come from sources, among others, including MGI (Blake et al. 2014), IMPC (Koscielny et al. 2014), ZFIN (Howe et al. 2013) and for human, OMIM (Amberger et al. 2015), ClinVar (Landrum et al. 2014), and Orphanet (Rath et al. 2012). For a number of sources we also perform extensive manual curation of gene-disease-phenotype associations (for example for human, see Köhler et al. 2014). We have constructed a data warehousing pipeline which regularly pulls data from these external sources and consolidates them into the integrated Monarch knowledgebase.
In contrast to other Matchmaker efforts, the underlying Monarch repository has to make use of a wider set of vocabularies than HPO or International Consortium for Human Phenotype Terminologies (ICHPT). Many of the terms in these vocabularies are inappropriate for or not complete enough for description of phenotypes in other species. Our approach therefore relies on an ontology-matching strategy. Previously we have shown how phenotype ontologies from human, mouse and more distant species such as zebrafish and C. elegans can be integrated together (Mungall et al. 2010; Köhler et al. 2013) through the use of multi-species organ system ontologies (Mungall et al. 2012; Haendel et al. 2014). These exploit functional analogies and evolutionary homologies across species - for example, a human bone marrow phenotype would be matched against a zebrafish ‘head kidney’ (A structure found in teleost fish such as Zebrafish that is distinct from the kidney and shares function with the mammalian bone marrow) phenotype based on homology and shared function between these tissues. Conversely, a ‘head kidney’ should not match to a human kidney despite the lexical match. This ontology also allows connection across levels of scale (for example, between Purkinje cells and the cerebellum) or based on shared developmental origins.
The Monarch inter-species phenotype matching algorithm has been previously described (Washington et al. 2009; Smedley et al. 2013) and is available as a Java standalone tool or webservices called OwlSim (http://owlsim.org). Figure 3 shows an illustrative example of how the algorithm works. Note that we never expect a model system to completely recapitulate the features of a human disease, and even when individual phenotypes are shared, the different modalities offered by a clinical setting and a laboratory setting means that these phenotypes are frequently observed at different levels of granularity. Our algorithms take account of this, and score according to closeness of an individual's phenotypic features weighted over the whole profile.
Figure 3. Patient matching against known diseases and model organisms.
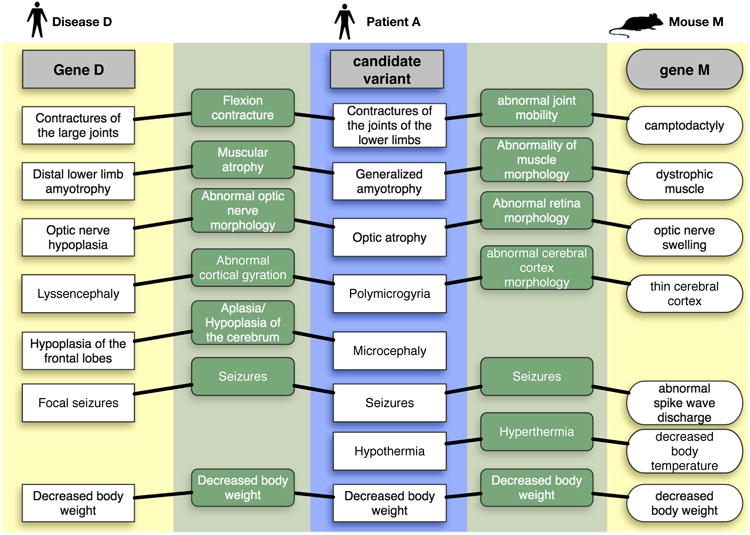
This figure illustrates matching from a patient to both a human disease and a mouse gene, using synthetic data. The center blue box represents the phenotypic profile of the undiagnosed disease patient encoded using HPO. The left yellow portion of the figure shows the closest matching known disease to this patient, where the disease information comes from public sources annotated with HPO by Monarch. On the right is a matching animal model, described using terms from the Mammalian Phenotype ontology (MPO). The green portions of the diagram shows the commonality between the matched phenotype terms both within species (left) and across species (right). Note that there are missing phenotypes in Disease D or Model M, another reason why comparison against the largest possible corpus is warranted. Patient A is based on a known undiagnosed disease patient that was solved based on phenotypic similarity to mouse and interactome data.
Even for moderately complex patients, matchmaking results can be significantly more complex than the example given in Figure 3. A given query with n input phenotypes might identify m candidate models, each of which having up to n phenotypes similar to the input profile. A moderately sized query potentially matching dozens of candidate models might lead to hundreds of phenotype similarities requiring interpretation. In our experience in developing Monarch tools, textual displays of these results have proven insufficient for critical tasks including comparisons of models, examination of individual phenotypes shared across multiple models, and identification of gaps – particularly in terms of phenotypes that are not recapitulated in otherwise similar models. To address these difficulties, we have developed the PhenoGrid visualization tool (Figure 4), which provides a compact tabular display of the pairwise similarity of phenotypic features between patients, diseases, and/or models. Currently deployed on the Monarch Initiative web site, PhenoGrid is available as a reusable web widget (http://www.github.com/monarch-initiative/phenogrid) that can be easily adapted and integrated into MME installations for the purposes of comparing patients, known diseases, and model organisms. Figure 4 illustrates a patient profile from the Undiagnosed Disease Program that prioritized a mutation in STIM1 (Markello et al 2015) based upon phenotypic similarity to a mouse mutant when combined with Exome data using the Exomiser tool (Robinson et al 2013).
Figure 4. Visualizing patient similarities to known diseases and model organisms.

An Undiagnosed Disease Program patient's phenotypes are on the left and match against the best genetic models in mice. The darker the square, the more in common the phenotypes are between the patient and the matched profile. Mouse models are shown here for comparison purposes. Note that a mouse mutant in the ortholog of STIM1 has 3 matching phenotypes with the patient's profile and when combined with exome analysis assisted the diagnosis of this patient. MME implementations of Phenogrid would enable comparison of input patient profiles against other patients accessible through MME protocols as well as known diseases and models.
Methods
Implementation of the MME API within Monarch
The implementation of the phenotype matching algorithm used by Monarch is called OwlSim. It is open source and can be installed in a variety of settings. OwlSim has its own APIs, both at the Java and REST levels, but this predates and is different from the MME API. In order to implement the MME API we created a bridge layer on top of our API to expose the services. This bridge is written in Scala using the Play framework (https://www.playframework.com/). A request to this MME implementation will be propagated to the OwlSim API, and the data coming back is transformed to match the MME API specifications before being sent to the user. The API can be queried on https://mme.monarchinitiative.org:9000.
Monarch and the GA4GH Ecosystem
The MME API has been adopted as part of the Global Alliance for Genomes & Health (GA4GH), an international coalition that aims to facilitate data sharing to advance human health. The GA4GH provides an integrated system of APIs (https://github.com/ga4gh/) that can be implemented and consumed by a variety of data and tool providers. The MME API is a part of this constellation of APIs, but has a distinct specification (https://github.com/MatchMakerExchange/mme-apis).
The Monarch consortium are participants in the GA4GH and are helping shape the APIs that address genotype to phenotype data exchange, both within the MME and in the Genotype-to-Phenotype (G2P) team. Figure 5 shows how Monarch fits into this ecosystem. We provide our own REST APIs that are highly tuned to some of the unique services we provide; all our data is modeled using rich semantic graphs, making use of the SciGraph framework (https://github.com/SciGraph/SciGraph). We implement the GA4GH APIs as an additional layer on top of this, providing a unified means of access to a broader range of tools and applications.
Figure 5. Monarch Architecture in the context of some other Global Alliance APIs.

such as the genotype-to-phenotype (G2P) API. The GA4GH APIs are implemented as a layer on top of our own REST services, which are backed by a SciGraph/Neo4J graph database.
Conclusions
Model systems can help diagnose disease and uncover novel disease-gene associations. The Monarch system provides a means of matching phenotypes between a human and a model organism. Through the standardized schemas of the MME, the Monarch Initiative contributes valuable knowledge for researchers, clinicians, and their IT personnel to integrate into MME-based interfaces, connecting model organism phenotypic resources and expertise with the clinic.
Acknowledgments
This work was supported by NIH under R24OD011883. CJM, NLW, JNX and SEL acknowledge the support of the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Funding: NIH under R24OD011883
Footnotes
The authors have no conflicts of interest.
References
- Amberger Joanna S, Bocchini Carol A, Schiettecatte François, Scott Alan F, Hamosh Ada. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Research. 2015 Jan 28;43(Database issue):D789–98. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake Judith A, Bult Carol J, Eppig Janan T, Kadin James A, Richardson Joel E. The Mouse Genome Database: Integration of and Access to Knowledge about the Laboratory Mouse. Nucleic Acids Research. 2014 Jan 26;42(Database issue):D810–7. doi: 10.1093/nar/gkt1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott Kym M, Vanstone Megan R, Bulman Dennis E, MacKenzie Alex E. Rare-Disease Genetics in the Era of next-Generation Sequencing: Discovery to Translation. Nature Reviews Genetics. 2013 Oct;14(10):681–91. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- Buske O, et al. The Matchmaker Exchange API: automating patient matching through the exchange of structured phenotypic and genotypic profiles. Hum Mutat. 2015;36:xxx–yyy. doi: 10.1002/humu.22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haendel Melissa A, Balhoff James P, Bastian Frederic B, Blackburn David C, Blake Judith A, Bradford Yvonne, Comte Aurelie, et al. Unification of Multi-Species Vertebrate Anatomy Ontologies for Comparative Biology in Uberon. Journal of Biomedical Semantics. 2014 Jan;5(1):21. doi: 10.1186/2041-1480-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe Douglas G, Bradford Yvonne M, Conlin Tom, Eagle Anne E, Fashena David, Frazer Ken, Knight Jonathan, et al. ZFIN, the Zebrafish Model Organism Database: Increased Support for Mutants and Transgenics. Nucleic Acids Research. 2013 Jan;41(Database issue):D854–60. doi: 10.1093/nar/gks938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettleborough Ross NW, Busch-Nentwich Elisabeth M, Harvey Steven A, Dooley Christopher M, Bruijn Ewart de, Eeden Freek van, Sealy Ian, et al. A Systematic Genome-Wide Analysis of Zebrafish Protein-Coding Gene Function. Nature. 2013 Apr 25;496(7446):494–7. doi: 10.1038/nature11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler Sebastian, Doelken Sandra C, Mungall Christopher J, Bauer Sebastian, Firth Helen V, Bailleul-Forestier Isabelle, Black Graeme CM, et al. The Human Phenotype Ontology Project: Linking Molecular Biology and Disease through Phenotype Data. Nucleic Acids Research. 2014 Jan;42(Database issue):D966–74. doi: 10.1093/nar/gkt1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler Sebastian, Doelken Sandra C, Ruef Barbara J, Bauer Sebastian, Washington Nicole, Westerfield Monte, Gkoutos George, et al. Construction and Accessibility of a Cross-Species Phenotype Ontology along with Gene Annotations for Biomedical Research. F1000Research. 2013 Jan;2:30. doi: 10.12688/f1000research.2-30.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscielny Gautier, Yaikhom Gagarine, Iyer Vivek, Meehan Terrence F, Morgan Hugh, Atienza-Herrero Julian, Blake Andrew, et al. The International Mouse Phenotyping Consortium Web Portal, a Unified Point of Access for Knockout Mice and Related Phenotyping Data. Nucleic Acids Research. 2014 Jan;42(Database issue):D802–9. doi: 10.1093/nar/gkt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum Melissa J, Lee Jennifer M, Riley George R, Jang Wonhee, Rubinstein Wendy S, Church Deanna M, Maglott Donna R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Research. 2014 Jan;42(Database issue):D980–5. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur Daniel G, Balasubramanian Suganthi, Frankish Adam, Huang Ni, Morris James, Walter Klaudia, Jostins Luke, et al. A Systematic Survey of Loss-of-Function Variants in Human Protein-Coding Genes. Science (New York, NY) 2012 Feb 17;335(6070):823–8. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markello T, Chen D, Kwan JY, Horkayne-Szakaly I, Morrison A, Simakova O, Gunay-Aygun M. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Molecular Genetics and Metabolism. 2015;114(3):474–82. doi: 10.1016/j.ymgme.2014.12.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall Christopher, Gkoutos Georgios, Smith Cynthia, Haendel Melissa, Lewis Suzanna, Ashburner Michael. Integrating Phenotype Ontologies across Multiple Species. Genome Biology. 2010;11(1):R2. doi: 10.1186/gb-2010-11-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall Christopher J, Torniai Carlo, Gkoutos Georgios V, Lewis Suzanna E, Haendel Melissa A. Uberon, an Integrative Multi-Species Anatomy Ontology. Genome Biology. 2012 Jan;13(1):R5. doi: 10.1186/gb-2012-13-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipakis Anthony, Azzariti Danielle, Beltra Sergi, et al. The Matchmaker Exchange: A Platform for Rare Disease Gene Discovery. Hum Mutat. 2015;36:xxx–yyy. doi: 10.1002/humu.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath Ana, Olry Annie, Dhombres Ferdinand, Brandt Maja Miličić, Urbero Bruno, Ayme Segolene. Representation of Rare Diseases in Health Information Systems: The Orphanet Approach to Serve a Wide Range of End Users. Human Mutation. 2012 May;33(5):803–8. doi: 10.1002/humu.22078. [DOI] [PubMed] [Google Scholar]
- Robinson Peter, Köhler Sebastian, Oellrich Anika, Wang Kai, Mungall Chris, Lewis Suzanna E, Washington Nicole, et al. Improved Exome Prioritization of Disease Genes through Cross Species Phenotype Comparison. Genome Research. 2013 Oct 25; doi: 10.1101/gr.160325.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson Peter N, Köhler Sebastian, Bauer Sebastian, Seelow Dominik, Horn Denise, Mundlos Stefan. The Human Phenotype Ontology: A Tool for Annotating and Analyzing Human Hereditary Disease. American Journal of Human Genetics. 2008 Nov;83(5):610–5. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson Peter N, Webber Caleb. Phenotype Ontologies and Cross-Species Analysis for Translational Research. PLoS Genetics. 2014 Apr;10(4):e1004268. doi: 10.1371/journal.pgen.1004268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley Damian, Oellrich Anika, Köhler Sebastian, Ruef Barbara, Westerfield Monte, Robinson Peter, Lewis Suzanna, Mungall Christopher. PhenoDigm: Analyzing Curated Annotations to Associate Animal Models with Human Diseases. Database : The Journal of Biological Databases and Curation. 2013 Jan;2013 doi: 10.1093/database/bat025. bat025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Cynthia L, Eppig Janan T. Expanding the Mammalian Phenotype Ontology to Support Automated Exchange of High Throughput Mouse Phenotyping Data Generated by Large-Scale Mouse Knockout Screens. Journal of Biomedical Semantics. 2015 Mar 25;6(1):11. doi: 10.1186/s13326-015-0009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum Mutat. 2015;36:xxx–yyy. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tifft Cynthia J, Adams David R. The National Institutes of Health Undiagnosed Diseases Program. Current Opinion in Pediatrics Mutat. 2014 Dec;26(6):626–33. doi: 10.1097/MOP.0000000000000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Slyke Ceri E, Bradford Yvonne M, Westerfield Monte, Haendel Melissa A. The Zebrafish Anatomy and Stage Ontologies: Representing the Anatomy and Development of Danio Rerio. Journal of Biomedical Semantics. 2014 Jan;5(1):12. doi: 10.1186/2041-1480-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington Nicole L, Haendel Melissa a, Mungall Christopher J, Ashburner Michael, Westerfield Monte, Lewis Suzanna E. Linking Human Diseases to Animal Models Using Ontology-Based Phenotype Annotation. PLoS Biology. 2009 Nov;7(11):e1000247. doi: 10.1371/journal.pbio.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein John N, Collisson Eric A, Mills Gordon B, Shaw Kenna R Mills, Ozenberger Brad A, Ellrott Kyle, Shmulevich Ilya, Sander Chris, Stuart Joshua M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nature Genetics. 2013 Oct;45(10):1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemojtel TS, Kohler L, Mackenroth M, Jager J, Hecht P, Krawitz L, Graul-Neumann, et al. Effective Diagnosis of Genetic Disease by Computational Phenotype Analysis of the Disease-Associated Genome. Science Translational Medicine. 2014 Sep 3;6(252):252ra123–252ra123. doi: 10.1126/scitranslmed.3009262. [DOI] [PMC free article] [PubMed] [Google Scholar]