




Abstract
Poly (ADP‐ribose) polymerase (PARP) inhibitors effectively kill tumours defective in the BRCA1 or BRCA2 genes through the concept of synthetic lethality. It is suggested that PARP inhibitors cause an increase in DNA single‐strand breaks (SSBs), which are converted during replication to irreparable toxic DNA double‐strand breaks (DSBs) in BRCA1/2 defective cells. There are a number of recent reports challenging this model. Here, alternative models that are not mutually exclusive are presented to explain the synthetic lethality between BRCA1/2 and PARP inhibitors. One such model proposes that PARP inhibition causes PARP‐1 to be trapped onto DNA repair intermediates, especially during base excision repair. This may in turn cause obstruction to replication forks, which require BRCA‐dependent homologous recombination to be resolved. In another model, PARP is directly involved in catalysing replication repair in a distinct pathway from homologous recombination. Experimental evidence supporting these novel models to explain the PARP‐BRCA synthetic lethality are discussed.
Keywords: Review, Homologous recombination, Stalled replication fork, DNA double-strand breaks, Poly(ADP-ribose) polymerase, BRCA1, BRCA2, Synthetic lethality, Cancer
Highlights
PARP‐1 is not a base excision repair protein.
SSBs do not accumulate as a primary lesion after PARP inhibition.
PARP is hyperactivated in BRCA2 defective cells, reactivating stalled forks.
1. Introduction
Inherited mutations in one copy of either the BRCA1 or BRCA2 gene is associated with a high risk of developing primarily breast and ovarian cancer (Miki et al., 1994; Wooster et al., 1995). Cancers arising in these individuals have lost a functional copy of BRCA1 or BRCA2. Hence, the BRCA1 and BRCA2 proteins are tumour suppressors and are required for homologous recombination (HR) to suppress genetic instability, which can lead to cancer (Venkitaraman, 2002). BRCA1 and BRCA2 defective tumours are intrinsically sensitive to PARP inhibitors, both in tumour models in vivo (Bryant et al., 2005; Evers et al., 2010; Farmer et al., 2005; Liu et al., 2007; Rottenberg et al., 2008) and in the clinic (Fong et al., 2009). Only mild side effects have been reported from PARP inhibitor treatment (Fong et al., 2009), which can be attributed to PARP inhibitors selectively targeting BRCA defective cells, owing to their defect in HR (Bryant et al., 2005; Farmer et al., 2005). Normal cells, with intact HR, are not significantly affected, in line with evidence that PARP‐1−/− mice are alive and healthy in general (de Murcia et al., 1997; Wang et al., 1997).
The genetic interaction between PARP and BRCA can be described as synthetic lethal. Synthetic lethality between two genes occurs where individual loss of either gene is compatible with life, but simultaneous loss of both genes results in cell death. It has for a long time been suggested that a synthetic lethal approach could be used in the treatment of cancer (Hartwell et al., 1997) and the PARP‐BRCA interaction provides the first example of a successful synthetic lethal approach that has entered the clinic.
Although several years have passed since the initial reports on the PARP‐BRCA synthetic lethality, we have so far not seen any other synthetic lethal approach reach the clinic. One possible reason for the slow pace in the development of new drugs using this concept may be our inability to mechanistically explain the PARP‐BRCA synthetic lethality. Indeed, mechanistic understanding has not been helped by the publication of numerous statements without support from the literature. Here, I will review recent findings that affect our mechanistic understanding of the PARP‐BRCA synthetic lethality.
2. PARP‐1 is not a base excision repair protein
It is well established that the PARP‐1 protein binds to SSBs, where it is activated to convert NAD+ into ADP‐ribose polymers (PAR), and that the protein is required for efficient SSB repair (Fisher et al., 2007; Satoh and Lindahl, 1992; Strom et al., 2011) by attracting XRCC1 to the site of damage (El‐Khamisy et al., 2003) (Figure 1A).
Figure 1.
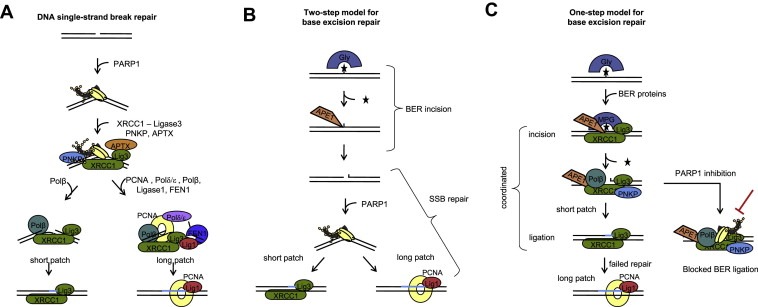
Base excision repair (BER) is a separate process from DNA single‐strand break (SSB) repair in mammalian cells, although the two processes share proteins. (A) SSB repair: PARP‐1 has a high affinity for SSBs and will be amongst the first proteins to bind to the lesion. In turn PARP recruits factors to start end processing and finally ligation, normally through short patch repair and through long patch repair where the lesions are more difficult to repair. (B) Two‐step model for BER: Different base lesions are recognised by different glycosylases (Gly), which are excised before SSB incision by the AP‐endonuclease (APE). These SSBs are then left unprotected and recognised in a separate process by PARP‐1 that will then initiate SSB repair. (C) One‐step model for BER: The glycosylase interacts with proteins involved in the early BER incision step and excises the damaged base shortly before APE incision. The half‐life of the SSB intermediate is very short and rapidly ligated by short patch repair, which switches to long patch repair in case of ligation difficulty. PARP‐1 has no role in BER, but can transiently bind the SSB intermediate. When PARP‐1 is inhibited, it can be trapped on the SSB intermediate and prevent the ligation step.
Traditionally, BER has been suggested to work as a series of independent steps, starting with removal of the damaged base, followed by separate recognition by AP‐endonuclease (APE), which makes a SSB incision. This unprotected SSB acts as a substrate for SSB repair (SSBR) involving PARP‐1 (Figure 1B). Indeed, PARP‐1 has been suggested to have a role in BER (Dantzer et al., 1999, 2000). This suggestion is well founded, as PARP‐inhibited or PARP‐1−/− cells are hypersensitive to agents that cause base lesions (de Murcia et al., 1997; Wang et al., 1997) and PARP‐1 is required for the rapid closure of alkylation‐induced SSBs (Trucco et al., 1998). Furthermore, ligation of AP‐sites generated from uracil or 8‐oxoguanine lesions is delayed in extracts from PARP‐1−/− cells. A potential caveat of these experiments is that damaged DNA and AP‐sites can be heat sensitive, which may cause these lesions to be converted into SSBs (Lundin et al., 2005). In addition, alkylated DNA bases effectively block replication elongation (Groth et al., 2010), and the sensitivity in PARP‐1−/− cells to those agents may be related to a role for PARP‐1 at replication forks (see below).
Other scientists have reported that BER kinetics are reduced in the presence of the active PARP‐1 protein (Allinson et al., 2003). Thus, the role of PARP‐1 in BER has remained elusive. Recently, we have set up an assay to measure BER incision in cells and the half‐life of the SSB intermediate formed during BER (Strom et al., 2011). Using this assay, we find that PARP‐1 is not required for BER in cells, but rather that the presence of PARP‐1 protein reduces the BER turnover (Strom et al., 2011). These data support a model where BER occurs in a single, coordinated step that is PARP‐1 independent (Figure 1C). Furthermore, these data suggest that SSBR is distinct from this process.
In the DNA repair literature, it has not been suggested that PARP‐1 is essential for BER, as such a notion would be ill founded. BER is an absolutely essential process and mouse APE1 (Xanthoudakis et al., 1996), Polβ (Gu et al., 1994) or XRCC1 (Tebbs et al., 2003) knockouts die at embryonic stages, in contrast to PARP‐1−/− mice (de Murcia et al., 1997; Wang et al., 1997). Repairing base lesions is essential for all life and the BER process is also highly conserved through evolution, in contrast to PARP proteins, which are poorly conserved and are not present in S. cerevisiae. Since some databases have annotated PARP‐1 to be a BER protein, there are now several papers in the literature stating that this is the case. I do hope that this notion can be revised, in light of currently available data.
Interestingly, we observe a complete opposite effect when using PARP inhibitors to deplete PARP‐1 rather than siRNA. These data demonstrate that PARP inhibitors inhibit BER by trapping the SSB intermediate, which may be an important contributor for selectively killing BRCA defective cells (Strom et al., 2011). Here, it is worth mentioning that BRCA defective cells are far more sensitive to PARP inhibitors than to siRNA knockdown of PARP‐1 (Bryant et al., 2005; Farmer et al., 2005). Hence, trapping PARP on specific DNA lesions, including BER intermediates, may be important for the effective killing of HR defective cells.
3. SSBs do not accumulate as a primary lesion after PARP inhibition
A short treatment (less than 24 h) with a PARP inhibitor is sufficient to trigger cell death in BRCA defective cells. Our originally proposed model suggested that PARP inhibition results in an increase in SSBs that then collapse replication forks into toxic one‐ended DSBs (Bryant et al., 2005; Farmer et al., 2005), which are substrates for HR (Arnaudeau et al., 2001; Helleday, 2003). Since BRCA defective cells are unable to perform HR, such toxic DSBs would accumulate in the cancer cells (Figure 2A). Although PARP inhibition delays repair of induced SSBs, the steady state level of SSBs is not increased with PARP inhibition in either wild type or BRCA2 defective cells, as observed using the alkaline comet assay (Gottipati et al., 2010). Furthermore, we cannot identify any SSBs after PARP inhibition or siRNA knockdown in undamaged cells using the sensitive alkaline DNA unwinding assay to detect SSBs (Strom et al., 2011). In addition, other laboratories report that background levels of SSBs appear to be normal in PARP‐1−/− or siRNA depleted cells (Fisher et al., 2007). Although there is a clear increase in γH2AX foci after PARP inhibition, there is no detection of any increased levels of DSBs as measured by pulsed‐field gel electrophoresis. This may also relate to the less sensitive method of pulsed‐field gel electrophoresis, which cannot distinguish fewer than 10 DSBs. Clearly, SSBs and DSBs will be formed after prolonged PARP inhibitor treatment, especially in BRCA defective cells, as cells enter apoptosis. Such DNA strand breaks are secondary and merely a consequence during cell death.
Figure 2.
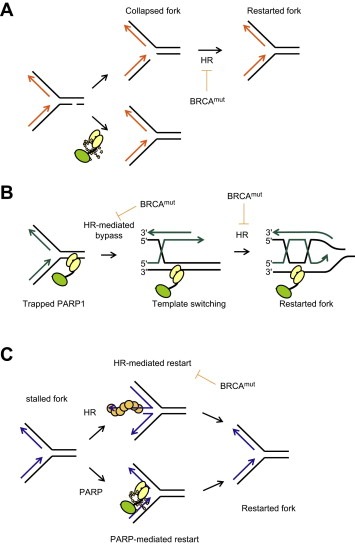
Pathways underlying PARP‐BRCA synthetic lethality. (A) SSB replication run‐off model. PARP‐1 is involved in repair of SSBs, which may in the presence of a PARP inhibitor persist and collapse a replication fork into a one‐ended DSB. Since BRCA defective cancer cells lack HR, the resulting DSBs would be selectively toxic to the cancer cells. (B) PARP‐1 trapping model. PARP inhibitors trap PARP‐1 onto SSBs formed spontaneously or as an intermediate during BER. Trapped PARP‐1 may pose an obstacle to replication that would require HR to bypass. (C) Replication restart model. In the case of normal replication, forks will stall owing to lack of replication factors or by obstacles on the DNA template. PARP and HR are activated at stalled forks and mediate distinct pathways for restart.
It is well established that an important role of PARP in SSBR is the recruitment of XRCC1 (Caldecott, 2003; El‐Khamisy et al., 2003). If the mechanism of PARP inhibitors is through deactivation of SSBR one would expect XRCC1 to also be synthetic lethal to BRCA1 or BRCA2. In contrast to this expectation, siRNA depletion of PARP‐1 but not XRCC1 reduces survival in BRCA2 defective cells (Patel et al., 2011), suggesting that the role of PARP‐1 in SSBR has little to do with the increased cell death in BRCA2 defective cells.
As mentioned earlier, PARP inhibitors trap a SSB intermediate during BER and delay SSBR. Hence, one may expect that the background levels of SSBs to increase upon exposure to a PARP inhibitor, as it would prevent repair of many of the SSBs arising normally in the cell every day (Lindahl, 1993). It is unclear why this is not the case, but may be explained by a slow turnover of repair and induction of SSBs and base lesions under normal conditions. Anyhow, since a PARP inhibitor may trap PARP onto the lesion or a DNA repair intermediate this complex may be converted to a more toxic lesion during replication (Figure 2B). This would be a similar mechanism of action as topoisomerase I poisons, such as camptothecin, which trap the topoisomerase I in a cleaved complex on the DNA, which is converted to a more toxic lesion during replication (Strumberg et al., 2000). Such a mechanism for PARP inhibitors could be possible in the context of an HR defective background, where it is suppressed in normal cells. The intermediate formed by trapping PARP to the DNA lesion is not comparable to the toxic lesions formed by camptothecin at replication forks, since trapping PARP is not particularly toxic to normal cells.
Another interesting aspect is that PARP inhibitors selectively kill XRCC1 defective cells (Strom et al., 2011) by an unknown mechanism. This is likely to be relevant to further understanding the PARP‐BRCA synthetic lethality.
4. Interaction between PARP‐1 and HR, a story of PARP inhibitors
The interaction between PARP‐1 and HR has been well documented for decades. Instrumental to our understanding of PARP‐1, we should acknowledge Sydney Shall, who very early on started to identify PARP inhibitors (Brightwell and Shall, 1971), paving the way for Barbara Durkacz in Dr Shall's research group to identify PARP as a DNA repair enzyme (Durkacz et al., 1980). Using the PARP inhibitors, an increase in sister chromatid exchanges (SCEs) following PARP inhibition was demonstrated (Morgan and Cleaver, 1982; Oikawa et al., 1980), which could later be verified in cells derived from PARP‐1 knockout mice (de Murcia et al., 1997; Wang et al., 1997). The role of PARP‐1 in controlling HR has for a long time been a major research area and it was for a long time suggested as a general recombination suppressor (Lindahl et al., 1995). In our hands, we found that the general increase in HR was unrelated to the HR process itself, demonstrating that PARP‐1 is not required for catalysing HR (Schultz et al., 2003). Although there has been a longstanding interest in the genetic interaction between HR and PARP inhibitors, it was only when more potent PARP inhibitors were synthesised that any translational aspect could be investigated (Griffin et al., 1995). Furthermore, the making of pharmacologically safe inhibitors, pioneered by KuDOS Ltd, was essential to make a difference in the clinic. In conclusion, the production of small molecule inhibitors of PARP was critical in identifying the role of PARP in DNA repair and as a target to selectively kill HR defective cancers.
5. PARP is hyperactivated in HR defective cells and has a role in reactivating stalled replication forks
The PARP‐1 protein has two very large DNA binding domains, with the ability to not only bind SSBs (Satoh and Lindahl, 1992), but also DNA double‐stranded ends as well as other DNA structures (D'Amours et al., 1999, 1999), including replication fork structures (Bryant et al., 2009). Interestingly, increased PARP activity is found in replicating cells (Lehmann et al., 1974) close to replication forks (Jump et al., 1979) and in newly replicated chromatin (Anachkova et al., 1989). Furthermore, PARP‐1 interacts with several DNA replication proteins, many of which are poly(ADP‐ribosyl)ated (Dantzer et al., 1998; Simbulan‐Rosenthal et al., 1996; Simbulan et al., 1993). Indeed, the PARP‐1 protein is activated at stalled replication forks and mediates effective restart of the stalled fork (Bryant et al., 2009; Yang et al., 2004). A similar function has also been demonstrated for several proteins involved in HR, e.g. RAD51, BLM, XRCC3 and FANCM (Davies et al., 2007; Petermann and Helleday, 2010; Petermann et al., 2010; Schwab et al., 2010). Since both PARP and HR proteins play an important role in restarting stalled replication forks, these functions may be important to understand the molecular mechanism for the synthetic lethality between PARP and BRCA proteins.
Interestingly, a recent publication demonstrates that BRCA2 protects from Mre11‐depedent degradation of stalled replication forks, a function unrelated to HR (Schlacher et al., 2011). BRCA2 protein unable to protect stalled replication forks still reverts to PARP inhibitor resistance, suggesting that this function may not be so important for the PARP‐BRCA synthetic lethality. However, our own observations demonstrate that PARP itself is critical to prevent from Mre11‐depedent degradation of stalled replication forks. Hence, the role of PARP and BRCA2 in both protecting from degradation of stalled forks is likely highly relevant to the mechanism of synthetic lethality.
Non‐homologous end joining (NHEJ) and DNA‐PK also play an important role at stalled replication forks (Lundin et al., 2002; Saintigny et al., 2001), which is so far poorly defined. Interestingly, the toxicity, chromosomal aberrations and mutations caused by PARP inhibitors in BRCA2 defective cells are suppressed by inhibition or loss of DNA‐PK (Patel et al., 2011). Since chromosomal aberrations and mutations are repaired during replication it is reasonable to believe that DNA‐PK influences replication repair in a way that prevents PARP inhibitors from being toxic in BRCA2 defective cells. These data suggest that DNA‐PK is first activated at replication forks to channel the repair through a pathway that involves PARP and BRCA. Hence, if DNA‐PK is inactive the repair will be channelled through a pathway that normally would not be used, but that now would circumvent the PARP and/or BRCA pathway. Similar data has also been reported in DT40 Chicken cells following treatments with camptothecin (Hochegger et al., 2006). Furthermore, recent findings showing that loss of 53BP1 in BRCA1 mutant cells alleviates hypersensitivity to PARP inhibitors and restores HR suggest a role for BRCA1 and 53BP1 in regulating the choice between HR and NHEJ pathways for DNA repair (Bouwman et al., 2010; Bunting et al., 2010).
We have previously reported that PARP‐1 is hyperactivated in BRCA2 and other HR defective cells (Gottipati et al., 2010). It is reasonable to believe that this hyperactivation of PARP is important to mediate survival in HR defective cells. The PARP‐1 hyperactivation is restricted to cells present in the S‐phase of the cell cycle, providing even more support for the presence of PARP to relieve or repair replication damage occurring in HR defective cells. I propose a model for the synthetic lethality that involves separate pathways for PARP and BRCA in mediating replication repair (Figure 2C) and is dependent on a functional NHEJ pathway.
In the clinic, some patients with confirmed BRCA mutations respond poorly to PARP inhibitors (Fong et al., 2009), while other ovarian cancers, with no apparent BRCA defect respond well to PARP inhibitor therapy (Gelmon et al., 2010). This highlights the need for predictive biomarkers to identify tumours that will respond to PARP inhibitor therapy. Since BRCA2 defective cells with acquired resistance to PARP inhibitors also revert to a low level of PARP activation (Gottipati et al., 2010), the measurement of PAR polymers (a readout of PARP activity) could be a useful biomarker to identify tumours that will respond to PARP inhibitor therapy. Alternative methods could be to determine RAD51 foci formation in tumours, an indicator of functional HR (Mukhopadhyay et al., 2010; Willers et al., 2009), or the presence of miRNAs such as miR‐182, which regulates the expression of the BRCA1 protein (Moskwa et al., 2011). In addition, 53BP1 expression in BRCA1 negative tumours could potentially be used to determine sensitivitiy to PARP inhibitors.
6. PARP inhibitors target PARP family members other than PARP‐1
Although most PARP inhibitors are developed to inhibit PARP‐1, most of them also inhibit PARP‐2, in some cases PARP‐3 (Farmer et al., 2005; Lehtio et al., 2009; Zaremba and Curtin, 2007), and potentially any of the other 16 members of the PARP superfamily (Schreiber et al., 2006).
The most evident role of PARP‐2 in suppressing the formation of spontaneous toxic lesions is the embryonic lethal phenotype observed in PARP‐1/PARP‐2 double knockout mice (Menissier de Murcia et al., 2003), strongly suggesting a role for PARP‐2 in some form of DNA repair. Although PARP‐2 has been suggested to play a role in BER together with PARP‐1 (Schreiber et al., 2002), other reports suggest only a minor role for PARP‐2 in SSBR (Fisher et al., 2007) and potentially a more important role in replication repair (Bryant et al., 2009).
PARP‐3 has recently been demonstrated to have a role together with APLF in accelerating NHEJ (Rulten et al., 2011), which also to involves PARP‐1 (Boehler et al., 2011). Other data show that PARP‐3 has a role in mitotic progression by associating and regulating NuMA and tankyrase 1(Boehler et al., 2011). Furthermore, there are robust protein interactions between PARP‐1, ‐2 and ‐3, which are of functional importance (Augustin et al., 2003; Loseva et al., 2010; Schreiber et al., 2002). For instance, PARP‐3 can activate PARP‐1 also in the absence of DNA (Loseva et al., 2010). Here, I have only briefly described some of the emerging roles of PARPs 1–3 in DNA repair, and we should expect to see more novel DNA repair roles catalysed by these or other PARP superfamily members uncovered in the not too distant future. Furthermore, PARPs have functions outside DNA repair in transcription, mitotic spindle formation, telomere cohesion, intracellular trafficking and energy metabolism (Schreiber et al., 2006), processes that may also contribute to the synthetic lethal phenotype between PARP and HR.
7. Conclusions and future directions
It is clear that the initial model, where PARP inhibitors cause replication‐associated DSBs by preventing SSBR and thereby killing HR defective cells, is incomplete. Additional factors important in understanding the synthetic lethality are: (1) the trapped PARP‐1 on the SSB intermediate during BER; (2) the role of PARP at stalled replication forks and (3) hyperactivated PARP in HR defective cells, as a likely result of accumulation of replication lesions in HR defective cells. Furthermore, given that PARP inhibitors target PARPs other than PARP‐1, which also have important functions for cell viability and DNA repair, we are likely to uncover new mechanisms for how PARP inhibitors mediate selective killing of HR defective cells.
Future research should be aimed at understanding the functions of the large PARP superfamily of proteins. As mentioned earlier, the success of PARP owes a debt of gratitude to the academic chemical biology approach and the making of pharmacologically active compounds by for instance KuDOS Ltd. In the future, selective inhibitors for different PARP family members should be developed to better be able to understand the roles of the individual PARP family members. The mechanism underlying the PARP‐BRCA synthetic lethality is clearly focussing on spontaneous damage arising at replication forks. A much stronger effort is needed to understand the nature of the lesions and the repair pathways present at replication forks. Increasing our understanding of the underlying mechanism for the PARP‐BRCA synthetic lethality is likely to help us to tailor novel synthetic lethal approaches also for other cancers.
Acknowledgements
I thank the The Swedish Cancer Society, the Swedish Children's Cancer Foundation the Swedish Research Council, the Swedish Pain Relief Foundation, the European Research Council and the Medical Research Council for funding.
Helleday Thomas, (2011), The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.07.001.
References
- Allinson, S.L. , Dianova, Dianov, G.L. , 2003. Poly(ADP-ribose) polymerase in base excision repair: always engaged, but not essential for DNA damage processing. Acta Biochim. Pol. 50, 169–179. [PubMed] [Google Scholar]
- Anachkova, B. , Russev, G. , Poirier, G.G. , 1989. DNA replication and poly(ADP-ribosyl)ation of chromatin. Cytobios. 58, 19–28. [PubMed] [Google Scholar]
- Arnaudeau, C. , Lundin, C. , Helleday, T. , 2001. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J. Mol. Biol.. 307, 1235–1245. [DOI] [PubMed] [Google Scholar]
- Augustin, A. , Spenlehauer, C. , Dumond, H. , Menissier-De Murcia, J. , Piel, M. , Schmit, A.C. , 2003. PARP-3 localizes preferentially to the daughter centriole and interferes with the G1/S cell cycle progression. J. Cell Sci.. 116, 1551–1562. [DOI] [PubMed] [Google Scholar]
- Boehler, C. , Gauthier, L.R. , Mortusewicz, O. , Biard, D.S. , Saliou, J.M. , Bresson, A. , 2011. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. U S A. 108, 2783–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman, P. , Aly, A. , Escandell, J.M. , Pieterse, M. , Bartkova, J. , van der Gulden, H. , 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol.. 17, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightwell, M. , Shall, S. , 1971. Poly(adenosine diphosphate ribose) polymerase in Physarum polycephalum nuclei. Biochem. J.. 125, 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, H.E. , Schultz, N. , Thomas, H.D. , Parker, K.M. , Flower, D. , Lopez, E. , 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose)polymerase. Nature. 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Bryant, H.E. , Petermann, E. , Schultz, N. , Jemth, A.S. , Loseva, O. , Issaeva, N. , 2009. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J.. 28, 2601–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting, S.F. , Callen, E. , Wong, N. , Chen, H.T. , Polato, F. , Gunn, A. , 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott, K.W. , 2003. XRCC1 and DNA strand break repair. DNA Repair (Amst). 2, 955–969. [DOI] [PubMed] [Google Scholar]
- D'Amours, D. , Desnoyers, S. , D'Silva, I. , Poirier, G.G. , 1999. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J.. 342, 249–268. [PMC free article] [PubMed] [Google Scholar]
- D'Silva, I. , Pelletier, J.D. , Lagueux, J. , D'Amours, D. , Chaudhry, M.A. , Weinfeld, M. , 1999. Relative affinities of poly(ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim. Biophys. Acta. 1430, 119–126. [DOI] [PubMed] [Google Scholar]
- Dantzer, F. , Nasheuer, H.P. , Vonesch, J.L. , de Murcia, G. , Menissier-de Murcia, J. , 1998. Functional association of poly(ADP-ribose) polymerase with DNA polymerase alpha-primase complex: a link between DNA strand break detection and DNA replication. Nucleic Acids Res.. 26, 1891–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer, F. , Schreiber, V. , Niedergang, C. , Trucco, C. , Flatter, E. , De La Rubia, G. , 1999. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 81, 69–75. [DOI] [PubMed] [Google Scholar]
- Dantzer, F. , de La Rubia, G. , Menissier-De Murcia, J. , Hostomsky, Z. , de Murcia, G. , Schreiber, V. , 2000. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 39, 7559–7569. [DOI] [PubMed] [Google Scholar]
- Davies, S.L. , North, P.S. , Hickson, I.D. , 2007. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol.. 14, 677–679. [DOI] [PubMed] [Google Scholar]
- de Murcia, J.M. , Niedergang, C. , Trucco, C. , Ricoul, M. , Dutrillaux, B. , Mark, M. , 1997. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. U S A. 94, 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkacz, B.W. , Omidiji, O. , Gray, D.A. , Shall, S. , 1980. (ADP-ribose)n participates in DNA excision repair. Nature. 283, 593–596. [DOI] [PubMed] [Google Scholar]
- El-Khamisy, S.F. , Masutani, M. , Suzuki, H. , Caldecott, K.W. , 2003. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res.. 31, 5526–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers, B. , Helleday, T. , Jonkers, J. , 2010. Targeting homologous recombination repair defects in cancer. Trends Pharmacol. Sci.. 31, 372–380. [DOI] [PubMed] [Google Scholar]
- Farmer, H. , McCabe, N. , Lord, C.J. , Tutt, A.N. , Johnson, D.A. , Richardson, T.B. , 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Fisher, A.E. , Hochegger, H. , Takeda, S. , Caldecott, K.W. , 2007. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol. Cell Biol.. 27, 5597–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong, P.C. , Boss, D.S. , Yap, T.A. , Tutt, A. , Wu, P. , Mergui-Roelvink, M. , 2009. Inhibition of Poly(ADP-ribose) polymerase in tumors from BRCA mutation Carriers. N. Engl. J. Med.. 361, 123–134. [DOI] [PubMed] [Google Scholar]
- Gelmon, K.A. , Hirte, H.W. , Robidoux, A. , Tonkin, K.S. , Tischkowitz, M. , Swenerton, K. , 2010. Can we define tumors that will respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serous ovarian cancer and triple-negative breast cancer. J. Clin. Oncol.. 28, 15s (suppl; abstr 3002) [Google Scholar]
- Gottipati, P. , Vischioni, B. , Schultz, N. , Solomons, J. , Bryant, H.E. , Djureinovic, T. , 2010. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res.. 70, 5389–5398. [DOI] [PubMed] [Google Scholar]
- Griffin, R.J. , Pemberton, L.C. , Rhodes, D. , Bleasdale, C. , Bowman, K. , Calvert, A.H. , 1995. Novel potent inhibitors of the DNA repair enzyme poly(ADP-ribose)polymerase (PARP). Anticancer Drug Des. 10, 507–514. [PubMed] [Google Scholar]
- Groth, P. , Auslander, S. , Majumder, M.M. , Schultz, N. , Johansson, F. , Petermann, E. , 2010. Methylated DNA causes a physical block to replication forks independently of damage signalling, O(6)-methylguanine or DNA single-strand breaks and results in DNA damage. J. Mol. Biol.. 402, 70–82. [DOI] [PubMed] [Google Scholar]
- Gu, H. , Marth, J.D. , Orban, P.C. , Mossmann, H. , Rajewsky, K. , 1994. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 265, 103–106. [DOI] [PubMed] [Google Scholar]
- Hartwell, L.H. , Szankasi, P. , Roberts, C.J. , Murray, A.W. , Friend, S.H. , 1997. Integrating genetic approaches into the discovery of anticancer drugs. Science. 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Helleday, T. , 2003. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res.. 532, 103–115. [DOI] [PubMed] [Google Scholar]
- Hochegger, H. , Dejsuphong, D. , Fukushima, T. , Morrison, C. , Sonoda, E. , Schreiber, V. , 2006. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. Embo J.. 25, 1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jump, D.B. , Butt, T.R. , Smulson, M. , 1979. Nuclear protein modification and chromatin substructure. 3. Relationship between poly(adenosine diphosphate) ribosylation and different functional forms of chromatin. Biochemistry. 18, 983–990. [DOI] [PubMed] [Google Scholar]
- Lehmann, A.R. , Kirk-Bell, S. , Shall, S. , Whish, W.J. , 1974. The relationship between cell growth, macromolecular synthesis and poly ADP-ribose polymerase in lymphoid cells. Exp. Cell Res.. 83, 63–72. [DOI] [PubMed] [Google Scholar]
- Lehtio, L. , Jemth, A.S. , Collins, R. , Loseva, O. , Johansson, A. , Markova, N. , 2009. Structural basis for inhibitor specificity in human poly(ADP-ribose) polymerase-3. J. Med. Chem.. 52, 3108–3111. [DOI] [PubMed] [Google Scholar]
- Lindahl, T. , 1993. Instability and decay of the primary structure of DNA. Nature. 362, 709–715. [DOI] [PubMed] [Google Scholar]
- Lindahl, T. , Satoh, M.S. , Poirier, G.G. , Klungland, A. , 1995. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci.. 20, 405–411. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Holstege, H. , van der Gulden, H. , Treur-Mulder, M. , Zevenhoven, J. , Velds, A. , 2007. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl. Acad. Sci. U S A. 104, 12111–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loseva, O. , Jemth, A.S. , Bryant, H.E. , Schuler, H. , Lehtio, L. , Karlberg, T. , 2010. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J. Biol. Chem.. 285, 8054–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin, C. , Erixon, K. , Arnaudeau, C. , Schultz, N. , Jenssen, D. , Meuth, M. , 2002. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell Biol.. 22, 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin, C. , North, M. , Erixon, K. , Walters, K. , Jenssen, D. , Goldman, A.S. , 2005. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res.. 33, 3799–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menissier de Murcia, J. , Ricoul, M. , Tartier, L. , Niedergang, C. , Huber, A. , Dantzer, F. , 2003. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. Embo J.. 22, 2255–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki, Y. , Swensen, J. , Shattuck-Eidens, D. , Futreal, P.A. , Harshman, K. , Tavtigian, S. , 1994. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 266, 66–71. [DOI] [PubMed] [Google Scholar]
- Morgan, W. , Cleaver, J. , 1982. 3-Aminobenzamide synergistically increases sister-chromatid exchanges in cells exposed to methyl methanesulfonate but not to ultraviolet light. Mutat. Res.. 104, 361–366. [DOI] [PubMed] [Google Scholar]
- Moskwa, P. , Buffa, F.M. , Pan, Y. , Panchakshari, R. , Gottipati, P. , Muschel, R.J. , 2011. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell. 41, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay, A. , Elattar, A. , Cerbinskaite, A. , Wilkinson, S.J. , Drew, Y. , Kyle, S. , 2010. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin. Cancer Res.. 16, 2344–2351. [DOI] [PubMed] [Google Scholar]
- Oikawa, A. , Tohda, H. , Kanai, M. , Miwa, M. , Sugimura, T. , 1980. Inhibitors of poly(adenosine diphosphate ribose) polymerase induce sister chromatid exchanges. Biochem. Biophys. Res. Commun.. 97, 1311–1316. [DOI] [PubMed] [Google Scholar]
- Patel, A.G. , Sarkaria, J.N. , Kaufmann, S.H. , 2011. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. U S A. 108, 3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann, E. , Helleday, T. , 2010. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol.. 11, 683–687. [DOI] [PubMed] [Google Scholar]
- Petermann, E. , Orta, M.L. , Issaeva, N. , Schultz, N. , Helleday, T. , 2010. Hydroxyurea-Stalled replication forks Become Progressively Inactivated and require two different RAD51-Mediated pathways for restart and repair. Mol. Cell. 37, 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg, S. , Jaspers, J.E. , Kersbergen, A. , van der Burg, E. , Nygren, A.O. , Zander, S.A. , 2008. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. U S A. 105, 17079–17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten, S.L. , Fisher, A.E. , Robert, I. , Zuma, M.C. , Rouleau, M. , Ju, L. , 2011. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell. 41, 33–45. [DOI] [PubMed] [Google Scholar]
- Saintigny, Y. , Delacote, F. , Vares, G. , Petitot, F. , Lambert, S. , Averbeck, D. , 2001. Characterization of homologous recombination induced by replication inhibition in mammalian cells. Embo J.. 20, 3861–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh, M.S. , Lindahl, T. , 1992. Role of poly(ADP-ribose) formation in DNA repair. Nature. 356, 356–358. [DOI] [PubMed] [Google Scholar]
- Schlacher, K. , Christ, N. , Siaud, N. , Egashira, A. , Wu, H. , Jasin, M. , 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber, V. , Ame, J.C. , Dolle, P. , Schultz, I. , Rinaldi, B. , Fraulob, V. , 2002. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem.. 277, 23028–23036. [DOI] [PubMed] [Google Scholar]
- Schreiber, V. , Dantzer, F. , Ame, J.C. , de Murcia, G. , 2006. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol.. 7, 517–528. [DOI] [PubMed] [Google Scholar]
- Schultz, N. , Lopez, E. , Saleh-Gohari, N. , Helleday, T. , 2003. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res.. 31, 4959–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, R.A. , Blackford, A.N. , Niedzwiedz, W. , 2010. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J.. 29, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simbulan, C.M. , Suzuki, M. , Izuta, S. , Sakurai, T. , Savoysky, E. , Kojima, K. , 1993. Poly(ADP-ribose) polymerase stimulates DNA polymerase alpha by physical association. J. Biol. Chem.. 268, 93–99. [PubMed] [Google Scholar]
- Simbulan-Rosenthal, C.M. , Rosenthal, D.S. , Hilz, H. , Hickey, R. , Malkas, L. , Applegren, N. , 1996. The expression of poly(ADP-ribose) polymerase during differentiation-linked DNA replication reveals that it is a component of the multiprotein DNA replication complex. Biochemistry. 35, 11622–11633. [DOI] [PubMed] [Google Scholar]
- Strom, C.E. , Johansson, F. , Uhlen, M. , Szigyarto, C.A. , Erixon, K. , Helleday, T. , 2011. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res.. 39, 3166–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumberg, D. , Pilon, A.A. , Smith, M. , Hickey, R. , Malkas, L. , Pommier, Y. , 2000. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell Biol.. 20, 3977–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbs, R.S. , Thompson, L.H. , Cleaver, J.E. , 2003. Rescue of Xrcc1 knockout mouse embryo lethality by transgene-complementation. DNA Repair (Amst). 2, 1405–1417. [DOI] [PubMed] [Google Scholar]
- Trucco, C. , Oliver, F.J. , de Murcia, G. , Menissier-de Murcia, J. , 1998. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res.. 26, 2644–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman, A.R. , 2002. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 108, 171–182. [DOI] [PubMed] [Google Scholar]
- Wang, Z.Q. , Stingl, L. , Morrison, C. , Jantsch, M. , Los, M. , Schulze-Osthoff, K. , 1997. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev.. 11, 2347–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willers, H. , Taghian, A.G. , Luo, C.M. , Treszezamsky, A. , Sgroi, D.C. , Powell, S.N. , 2009. Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol. Cancer Res.. 7, 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooster, R. , Bignell, G. , Lancaster, J. , Swift, S. , Seal, S. , Mangion, J. , 1995. Identification of the breast cancer susceptibility gene BRCA2. Nature. 378, 789–792. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis, S. , Smeyne, R.J. , Wallace, J.D. , Curran, T. , 1996. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. U S A. 93, 8919–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y.G. , Cortes, U. , Patnaik, S. , Jasin, M. , Wang, Z.Q. , 2004. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene. 23, 3872–3882. [DOI] [PubMed] [Google Scholar]
- Zaremba, T. , Curtin, N.J. , 2007. PARP inhibitor development for systemic cancer targeting. Anticancer Agents Med. Chem.. 7, 515–523. [DOI] [PubMed] [Google Scholar]