




Abstract
Systemic inflammatory stimuli, such as infection, increase the risk of stroke and are associated with poorer clinical outcome. The mechanisms underlying the impact of systemic inflammatory stimuli on stroke are not well defined. We investigated the impact of systemic inflammation on experimental stroke and potential mechanisms involved. Focal cerebral ischemia was induced by intraluminal filament occlusion of the middle cerebral artery (MCAo). Brain damage and neurological deficit 24 h after MCAo were exacerbated by systemic lipopolysaccharide (LPS) administration. This exacerbation was critically dependent on interleukin (IL)-1, because coadministration of IL-1 receptor antagonist abolished the effect of LPS on brain damage. Systemic administration of IL-1 increased ischemic damage to a similar extent as LPS and also exacerbated brain edema. IL-1 markedly potentiated circulating levels of the acute phase proteins, serum amyloid A and IL-6, and the neutrophil-selective CXC chemokines, KC and macrophage inflammatory protein-2. Neutrophil mobilization and cortical neutrophil infiltration were aggravated by IL-1 before changes in ischemic damage. Neutropenia abolished the damaging effects of systemic IL-1. These data show for the first time that an acute systemic inflammatory stimulus is detrimental to outcome after experimental stroke and highlight IL-1 as a critical mediator in this paradigm. Our data suggest IL-1-induced potentiation of neutrophil mobilization via CXC chemokine induction is a putative mechanism underlying this effect. Our results may help to explain the poorer outcome in stroke patients presenting with infection and may have implications for neurodegenerative diseases involving neurovascular alterations, such as Alzheimer's disease, in which systemic inflammation can modulate disease progression.
Keywords: cerebral ischemia, cytokine, infection, inflammation, leukocyte, lipopolysaccharide
Introduction
It is well established that local inflammatory processes in the brain contribute to the damage caused by acute brain injury and the progression of neurodegenerative disease. In contrast, the role of systemic inflammatory processes has been poorly investigated, although growing evidence suggests that the incidence of, and response to, acute brain injury and the progression of several neurodegenerative diseases are modulated by systemic inflammatory events such as infection (Emsley and Tyrrell, 2002; Perry, 2004). In particular, considerable clinical data indicate that acute and chronic systemic infection are risk factors for stroke and are associated with less favorable clinical outcome (Grau et al., 1995a,b; Smeeth et al., 2004; Palasik et al., 2005). The mechanisms underlying the effects of systemic inflammation on CNS injury are poorly defined.
The induction of systemic innate immune responses after stroke that also occur during the host defense response to infection may serve as a point of convergence when these insults are coincident. Stroke elicits a systemic acute phase response and is associated with increased levels of circulating cytokines, acute phase reactants, and elevated neutrophil and total leukocyte counts, which correlate with poor clinical outcome (Muir et al., 1999; Emsley et al., 2003; Smith et al., 2004; Waje-Andreassen et al., 2005; Rallidis et al., 2006). A recent study further highlighted the marked activation of systemic immune pathways after experimental stroke, including induction of cytokines and chemokines in peripheral lymphoid organs (Offner et al., 2006).
The proinflammatory cytokine, interleukin (IL)-1, is an important mediator of acute brain injury (Allan et al., 2005) and a key regulator of inflammation during the host defense response. IL-1 mediates several of the physiological and behavioral changes in response to a systemic inflammatory challenge induced by the bacterial endotoxin, lipopolysaccharide (LPS), or turpentine (Long et al., 1990; Luheshi et al., 1996, 1997; Miller et al., 1997b). IL-1 may act by inducing the expression of an array of downstream mediators, several of which are pertinent to the systemic inflammatory response after stroke, in particular IL-6 and chemokines (Miller et al., 1997b; Cartmell et al., 2000; Calkins et al., 2002). Treatment of acute stroke patients with the IL-1 receptor antagonist (IL-1RA) markedly attenuates the elevated plasma IL-6 and C-reactive protein levels and neutrophil counts (Emsley et al., 2005).
The objective of the present study was to investigate the impact of systemic inflammation on experimental stroke and to assess the role of IL-1 and downstream mediators in this context. We first show that peripheral LPS challenge exacerbates brain injury after focal cerebral ischemia, an effect critically dependent on IL-1. We then demonstrate that peripheral IL-1 challenge has pathological effects similar to LPS, acting via the induction of neutrophil-selective CXC chemokines and a neutrophil-dependent mechanism.
Materials and Methods
Mice.
All experiments were performed on 10- to 12-week-old (25–30 g) C57BL/6J mice (Harlan-Olac, Bicester, UK) under appropriate United Kingdom Home Office personal and project licenses and adhered to regulations as specified in the Animals (Scientific Procedures) Act (1986).
Focal cerebral ischemia.
Focal ischemia was induced by transient (30 min) middle cerebral artery occlusion (MCAo) using a modification of the intraluminal filament technique originally described (Longa et al., 1989). Core body temperature was regulated at 37 ± 0.5°C throughout the procedure by a feedback-controlled heating blanket. Under halothane anesthesia (30% O2/70% N2O), the carotid arteries were exposed and a 6-0 nylon monofilament (Dermalon) with 2 mm tip (180 μm diameter) coated in thermo-melting glue (Jet Melt) was introduced into the external carotid artery and advanced 9 mm along the internal carotid artery (ICA) until occluding the origin of the MCA. After 30 min, the filament was withdrawn to establish reperfusion. Sham-operated mice underwent the same procedure except the filament was advanced along the ICA, and then immediately withdrawn. Mortality rate was <15% in all groups except where stated.
Drug administration.
All treatments were administered in a blinded manner by intraperitoneal injection in a volume of 100 μl/25 g. LPS serotype 0127:B8 (Sigma, St. Louis, MO; 100 μg/kg in 0.9% sterile saline) or vehicle (0.9% sterile saline) was administered 30 min before the onset of MCAo. Recombinant human IL-1RA [National Institute for Biological Standards and Controls (NIBSC), Potters Bar, Hertfordshire, UK; 100 mg/kg in saline] or vehicle were coadministered with LPS 30 min before MCAo, and additional injections were given 1 and 4 h after MCAo. Recombinant IL-1β (NIBSC; biological activity, 100,000 IU/μg) diluted in vehicle (0.5% endotoxin-free bovine serum albumin in sterile PBS) at indicated doses, or vehicle was administered at the onset of MCAo. To deplete neutrophils, mice received three injections of rabbit anti-polymorphonuclear leukocyte (PMN) IgG (Accurate Scientific, Westbury, NY; 2 mg/kg) or rabbit IgG as control at the same dose diluted in sterile saline. One injection was given per day for 3 d before MCAo.
Assessment of neurological deficit.
Neurological status was assessed blinded to drug treatment and according to a neurological grading score of increasing severity of deficit (Bederson et al., 1986): 0, no observable deficit; 1, torso flexion to right; 2, spontaneous circling to right; 3, leaning/falling to right; 4, no spontaneous movement.
Tissue processing.
For measurement of ischemic damage and cerebral edema, brains were removed and snap-frozen in chilled isopentane. For immunohistochemistry, mice were perfused transcardially with 0.9% saline followed by 4% paraformaldehyde. Brains were removed, postfixed, cryoprotected (15% sucrose), and frozen. For all experiments, sections (20 μm) were cut on a cryostat (Leica Microsystems, Nussloch, Germany) and stored at −20°C. Plasma was extracted from anticoagulated (sodium citrate) blood samples obtained by cardiac puncture.
Measurement of ischemic brain damage and edema.
Coronal brain sections at 400 μm intervals were stained with cresyl violet (Sigma). Areas of ischemic damage at each coronal level were delineated and the volume of ischemic damage was derived from the sum of all areas of damage multiplied by the distance between each section (0.4 mm). The volume of damage was corrected for edema as described previously (Lin et al., 1993). The extent of edema was calculated by subtracting the volume of the hemisphere contralateral to MCAo from the volume of the ipsilateral hemisphere and expressing the difference as a percentage of the contralateral hemisphere. Ischemic damage and edema were measured blinded to drug treatment.
Circulating leukocyte counts.
Terminal cardiac blood samples were centrifuged at 700 × g and plasma aspirated. Erythrocytes were lysed and a leukocyte differential determined from Wright–Giemsa-stained smears.
Immunohistochemistry.
Endogenous peroxidase activity was blocked with 0.3% H2O2 in methanol and nonspecific binding sites were blocked with 10% normal serum (Vector Laboratories, Burlingame, CA). Sections were incubated in primary antibody solution [1:500 anti-neutrophil serum (MBS-1) (gift from V. H. Perry, University of Southampton, Southampton, UK) in 5% blocking solution] overnight at 4°C followed by appropriate biotinylated secondary antibody (Vector; 1:200 in PBS). Primary antibody incubation was omitted for assessment of blood–brain barrier (BBB) disruption using biotinylated anti-mouse IgG. Sections were then incubated in Vectastain ABC solution (Vector) and color was developed by diaminobenzidine incubation (Vector). Cerebral neutrophil infiltration was quantified in a blinded manner by counting the number of MBS-immunopositive cells in three areas of the cortex (somatosensory, insular, piriform) or striatum (dorsal, lateral, ventral) at two coronal levels (0.2 and −0.5 mm relative to bregma). The mean was calculated from the six fields in the cortex or striatum and adjusted to express as mean number of cells per square millimeter.
ELISA.
Plasma IL-6, KC, macrophage inflammatory protein-2 (MIP-2) (Duoset; R & D Systems, Minneapolis, MN), and serum amyloid A (SAA) (Biosource, Camarillo, CA) concentrations were determined by ELISA according to manufacturer's instructions.
Statistical analysis.
For all analyses, n = 6–8, except where stated. Parametric data were analyzed using Student's t test for single comparisons and one-way ANOVA followed by Student's t test with Bonferroni's correction or Dunnett's test for multiple comparisons. Nonparametric data were analyzed using Kruskal–Wallis test followed by Dunn's test for multiple comparisons.
Results
Acute systemic inflammatory challenge worsens outcome after experimental stroke
To determine the effect of a systemic inflammatory stimulus on acute ischemic brain damage, we administered bacterial endotoxin (LPS) intraperitoneally 30 min before MCAo and assessed the extent of ischemic damage and neurological deficit 24 h post-MCAo. LPS caused a 150% increase in the volume of ischemic damage compared with vehicle treatment (p < 0.01) (Fig. 1A), which was mostly attributable to exacerbation of cortical damage (Fig. 1B), and significantly increased the severity of neurological deficit (p < 0.05) (Fig. 1C).
Figure 1.
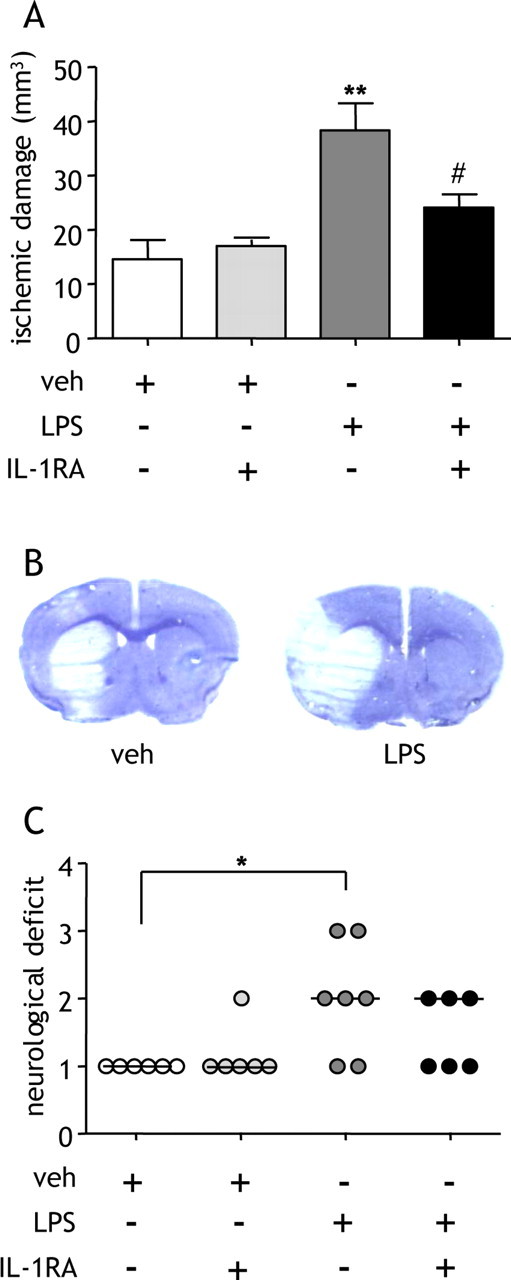
Systemic LPS exacerbates ischemic brain damage via an IL-1-dependent pathway. LPS was administered systemically, and the extent of ischemic damage and neurological deficit was assessed 24 h after MCAo. A, LPS significantly increased the volume of ischemic damage. Coadministration of IL-1RA attenuated the effect of LPS. **p < 0.01 versus vehicle (veh); #p < 0.05 versus LPS, one-way ANOVA followed by Student's t test with Bonferroni's correction. Data are presented as mean ± SEM. B, Representative cresyl violet-stained sections illustrate the distribution of ischemic damage after MCAo in vehicle- or LPS-treated mice. C, LPS worsened neurological outcome and coadministration of IL-1RA reduced the number of mice with a severe neurological deficit (score, 3). *p < 0.05 versus vehicle, Kruskal–Wallis test followed by Dunn's multiple-comparison test. Bars show median. n = 6–7 per group.
LPS induces several mediators, including IL-1, via activation of Toll-like receptors (TLRs). We tested the hypothesis that IL-1 mediates the effects of LPS on ischemic brain damage by coadministering LPS with the specific IL-1RA. Coadministration of IL-1RA significantly attenuated (p < 0.05) the LPS-induced exacerbation of damage by 60% and improved neurological status (Fig. 1A). To further examine the specific role of systemic IL-1 in the absence of potentially diverse and confounding actions of LPS, recombinant IL-1β was administered intraperitoneally and ischemic damage and neurological deficit were assessed 24 h after MCAo. IL-1β (10 or 100 IU) significantly exacerbated (>150%) the volume of ischemic damage compared with vehicle treatment (p < 0.05) (Fig. 2A) and significantly increased the severity of neurological deficit (10 IU; p < 0.05) (Fig. 2B). Mortality rate was >60% in mice treated with the high dose (100 IU) of IL-1β. All subsequent experiments were conducted using the lower dose (10 IU) of IL-1β, which is similar to the concentration and biological activity of IL-1β induced locally by LPS (100 μg/kg) injection (Miller et al., 1997a; Cartmell et al., 2000). Ischemic damage was primarily striatal in distribution in vehicle-treated mice, with IL-1β (10 IU) increasing ischemic damage predominantly in the cerebral cortex (p < 0.01) (Fig. 2C).
Figure 2.
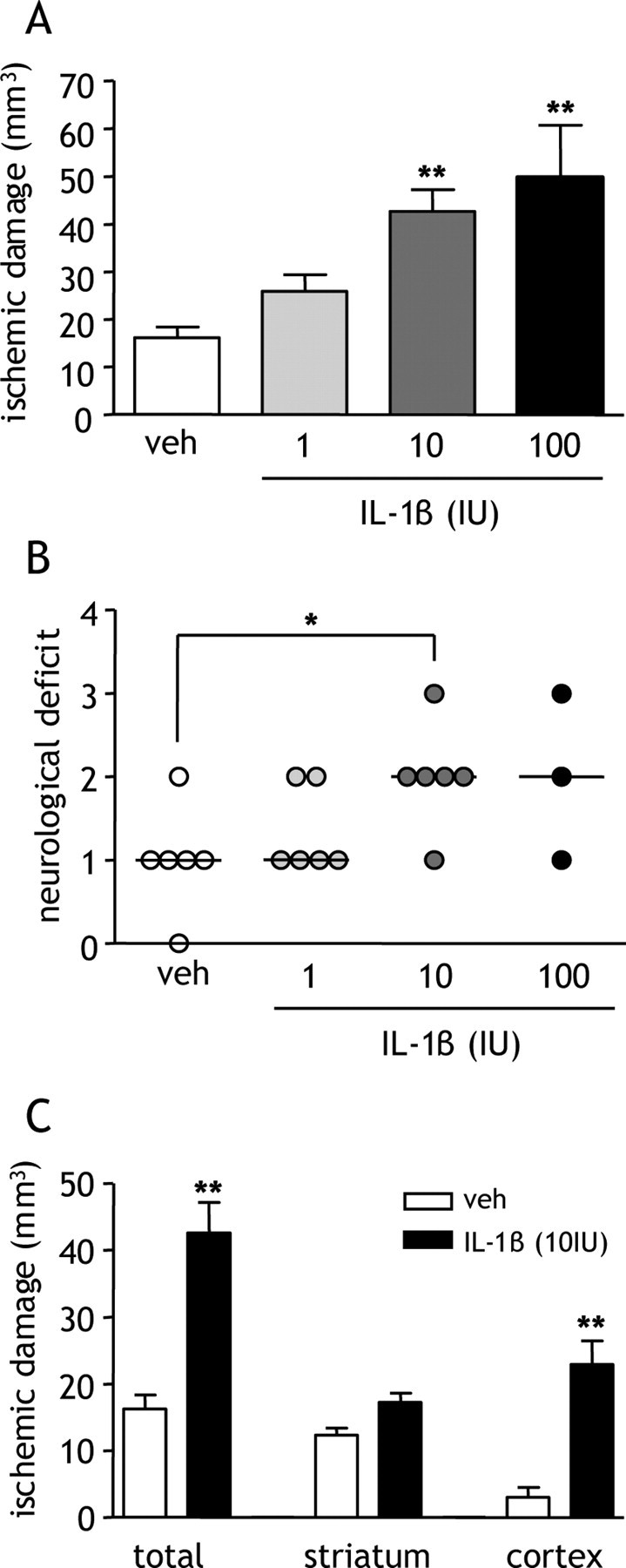
Dose-dependent exacerbation of ischemic brain damage by systemic IL-1β. Recombinant IL-1β was administered systemically at the onset of MCAo, and the extent of ischemic damage and neurological deficit was assessed 24 h after MCAo. A, IL-1β induced a dose-dependent exacerbation of ischemic damage. **p < 0.01 versus vehicle (veh), one-way ANOVA followed by Dunnett's multiple-comparison test. Data are presented as mean ± SEM. B, Neurological deficit was exacerbated in a dose-dependent manner by IL-1β. *p < 0.05 versus vehicle, Kruskal–Wallis test followed by Dunn's multiple-comparison test. Bars show median. C, IL-1β exacerbated damage in cerebral cortex but not in the striatum. **p < 0.01 versus vehicle, Student's t test. Data are presented as mean ± SEM; n = 6 per group except for IL-1β (100 IU)-treated group (n = 3) because of high mortality rate (>60%).
Systemic IL-1β exacerbates brain edema and blood–brain barrier permeability before ischemic damage
We next determined the effects of a systemic IL-1β challenge on the temporal progression of ischemic damage, edema, and BBB breakdown. There were no differences in the extent of ischemic damage 4 and 8 h after MCAo when damage was confined to the striatum after vehicle or IL-1β treatment (Fig. 3A). There was clear infarction in striatal and cortical tissue 24 h after MCAo, and IL-1β treatment resulted in a marked increase in the extent of damage (p < 0.01) (Fig. 3A), consistent with the data shown in Figure 1.
Figure 3.
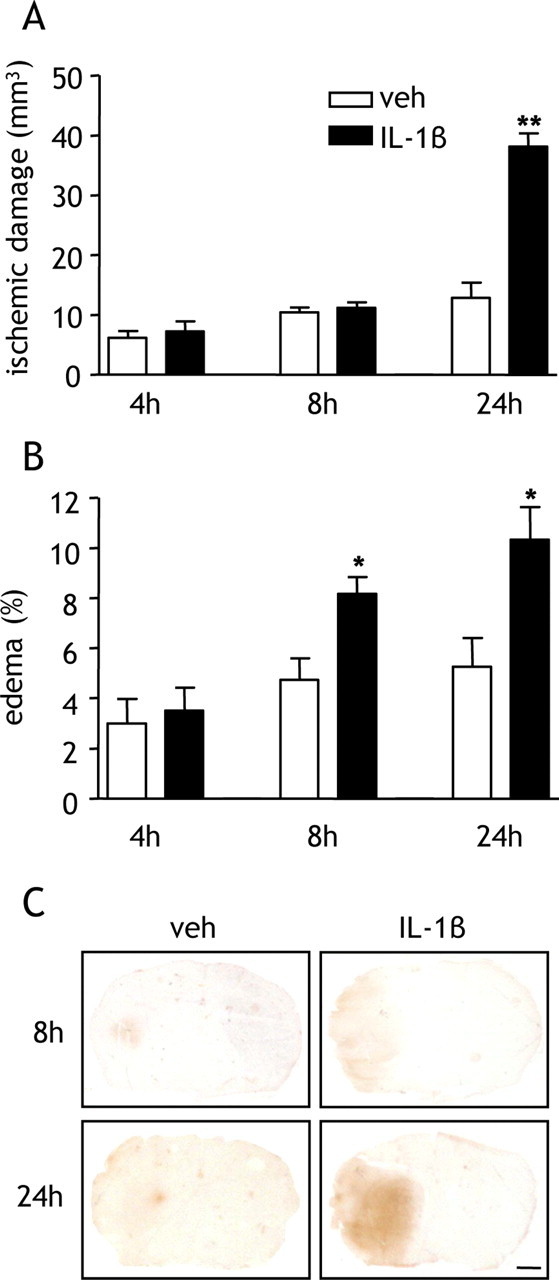
Systemic IL-1β potentiates brain edema and blood–brain barrier permeability after MCAo. Recombinant IL-1β was administered systemically at the onset of MCAo, and the temporal progression of ischemic damage, brain edema, and BBB disruption were assessed. A, IL-1β significantly exacerbated ischemic damage 24 h after MCAo but not 4 and 8 h after MCAo. B, IL-1β significantly increased the extent of edema 8 and 24 h after MCAo. C, Representative brain sections immunostained for IgG. IL-1β markedly increased the intensity and distribution of IgG immunoreactivity 8 and 24 h after MCAo, indicating potentiation of BBB disruption by IL-1β. *p < 0.05, **p < 0.01 versus vehicle (veh), Student's t test. Data are presented as mean ± SEM. Scale bar, 1 mm. n = 6–8 per group.
Edema was evident in the occluded hemisphere in both vehicle- and IL-1β-treated mice, and the extent of edema increased as reperfusion progressed (Fig. 3B). IL-1β significantly exacerbated edema 8 and 24 h after reperfusion (p < 0.05). Effects of IL-1β on edema were evident, therefore, before overt alterations in ischemic damage.
Increased permeability of the BBB contributes to vasogenic brain edema. We therefore assessed whether IL-1β exacerbated BBB disruption using IgG immunohistochemistry. Parenchymal IgG immunoreactivity reflects areas of disrupted BBB because IgG cannot cross the intact BBB. Marked increases in the distribution and intensity of IgG immunoreactivity were evident in IL-1β-treated mice 8 and 24 h after MCAo (Fig. 3C), suggesting IL-1β-induced escalation of early and ongoing BBB disruption after cerebral ischemia. Such an early exacerbation of BBB permeability is consistent with a causative role in the development of ischemic damage. There was no IgG immunostaining of brain tissue in sham-operated mice after vehicle or IL-1β treatment.
Systemic IL-1β potentiates the postischemic acute phase response
The temporal profile of the IL-1β-induced exacerbation of damage indicated the involvement of mechanisms progressing over several hours, consistent with the induction of peripheral inflammatory processes. We next determined whether a systemic IL-1β challenge affected the magnitude of the postischemic acute phase response, a key indicator of the extent of peripheral inflammation and highly correlative with prognosis in stroke patients. IL-1β greatly potentiated the plasma concentration of the acute-phase reactant, SAA, 4 h after MCAo, inducing a significant eightfold increase in SAA compared with vehicle treatment (Fig. 4A). The magnitude of the increase 4 h after MCAo indicated synergism between the ischemic and IL-1β challenges. SAA levels declined as reperfusion progressed. Plasma IL-6 levels were markedly increased 4 h after MCAo and declined as reperfusion progressed (Fig. 4B). IL-1β potentiated the plasma IL-6 concentration at all time points after MCAo, and these differences were statistically significant 4 and 8 h after MCAo (p < 0.05) (Fig. 4B). The magnitude of the IL-1β-induced elevations in plasma IL-6 after MCAo indicated synergistic effects between IL-1β and ischemic challenges.
Figure 4.
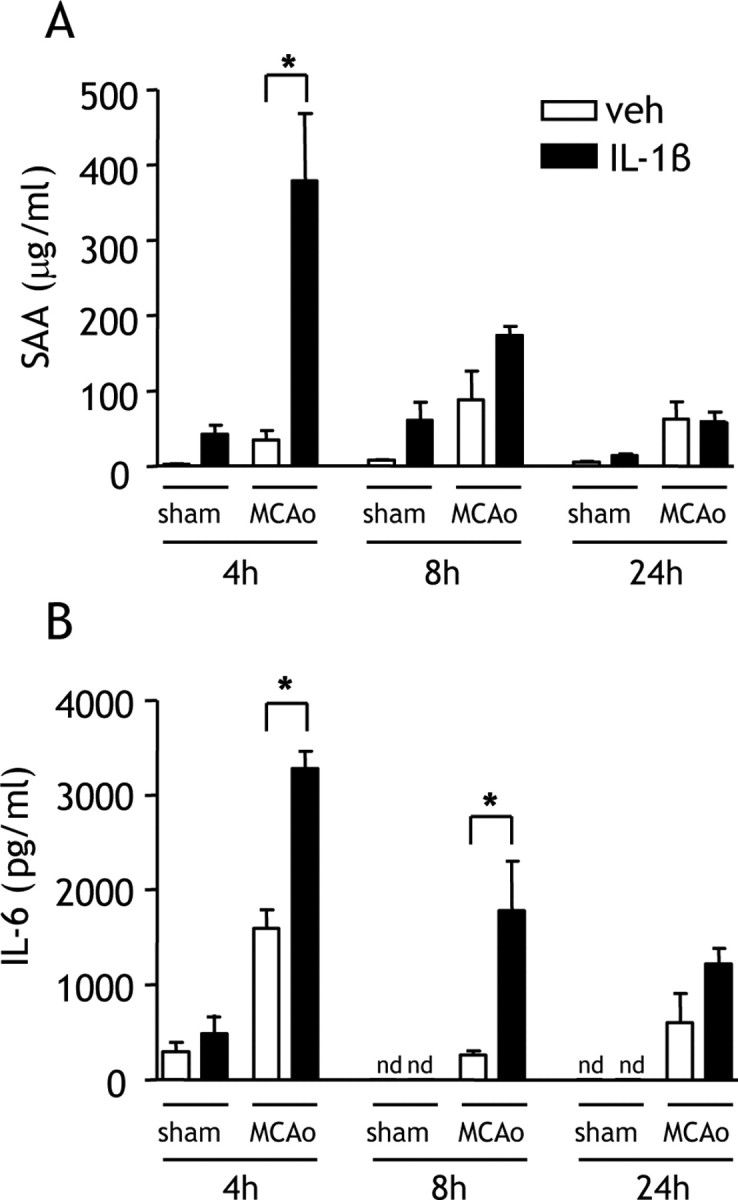
Systemic IL-1β exacerbates the acute-phase response after MCAo. Plasma SAA and IL-6 concentrations were measured by ELISA in mice administered vehicle (veh) or IL-1β systemically at the onset of MCAo. A, IL-1β significantly increased the concentration of plasma SAA 4 h after MCAo. B, IL-1β markedly increased the concentration of plasma IL-6 at all time points after MCAo, although the change was statistically significant only 4 h after MCAo. nd, Below detection limit. *p < 0.05 versus vehicle, Student's t test. Data are presented as mean ± SEM. n = 6–8 per group.
Systemic IL-1β potentiates postischemic neutrophilia and cortical neutrophil infiltration
Neutrophils respond to injury and/or infection by migrating into inflamed tissue within hours of the insult. We next tested whether a systemic IL-1β challenge potentiated the postischemic mobilization of neutrophils and their infiltration into the brain. In vehicle-treated mice, MCAo induced an increase in the number of circulating PMNs by over 30% compared with the preischemic value 8 and 24 h after MCAo with the peak neutrophilia 8 h after MCAo (Fig. 5A, Table 1). IL-1β treatment significantly potentiated the number of circulating PMNs 8 h after MCAo (p < 0.05) and to a lesser extent 4 and 24 h after MCAo. IL-1β did not significantly alter the number of circulating monocytes or lymphocytes (Table 1).
Figure 5.
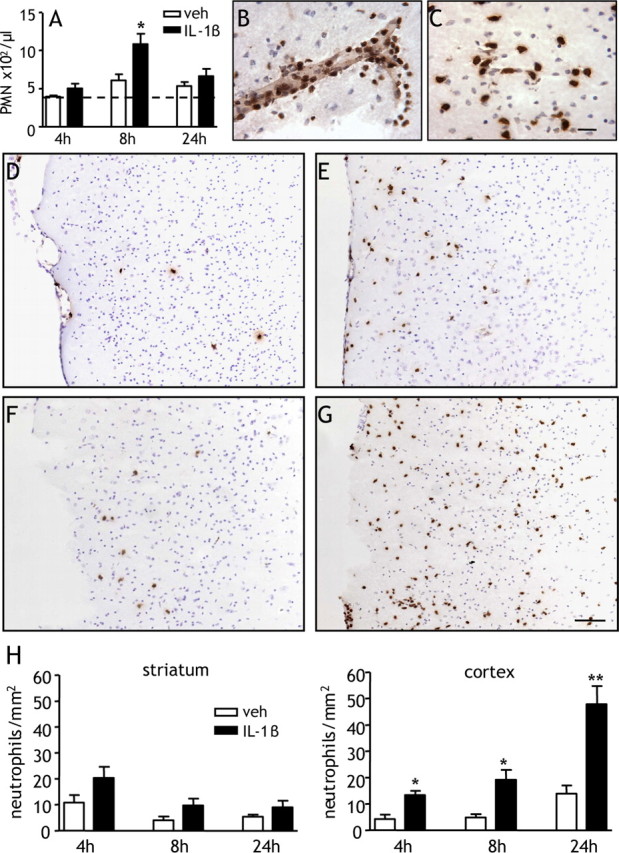
Systemic IL-1β potentiates neutrophilia and cortical neutrophil infiltration after MCAo. A, The number of circulating PMNs was assessed in mice administered vehicle (veh) or IL-1β systemically at the onset of MCAo. IL-1β significantly increased the number of circulating PMNs 8 h after MCAo. The dotted line indicates pre-MCAo value (vehicle and IL-1β groups pooled). B–D, Accumulation of neutrophils in the brain was assessed by anti-MBS-1 immunohistochemistry. Neutrophils were observed in the cerebral vasculature (B) and brain parenchyma (C). Representative brain sections illustrate neutrophil accumulation in cerebral cortex ipsilateral to MCAo 8 h (D, E) and 24 h (F, G) after MCAo. Marked increases in the number of neutrophils infiltrating into cortical tissue were observed in IL-1β-treated mice (E, G) compared with vehicle-treated mice (D, F). H, IL-1β significantly increased the number of neutrophils accumulating in the cerebral cortex 4, 8, and 24 h after MCAo. *p < 0.05, **p < 0.01 versus vehicle, Student's t test. Data are presented as mean ± SEM. Scale bars: A–D, 100 μm; E–G, 25 μm. n = 6–8 per group.
Table 1.
Effect of systemic IL-1β on circulating white blood cells after MCAo
| Pre-MCAoa | 4 h |
8 h |
24 h |
||||
|---|---|---|---|---|---|---|---|
| Vehicle | IL-1β | Vehicle | IL-1β | Vehicle | IL-1β | ||
| Total WBCs/μl | 2228 ± 117 | 1800 ± 105 | 1900 ± 148 | 1967 ± 229 | 2586 ± 385 | 1988 ± 214 | 1825 ± 43 |
| PMNs/μl | 380 ± 23 | 396 ± 12 | 501 ± 61 | 605 ± 80 | 1081 ± 138* | 531 ± 53 | 661 ± 95 |
| Monocytes/μl | 126 ± 7 | 115 ± 11 | 134 ± 18 | 141 ± 28 | 160 ± 13 | 216 ± 16 | 215 ± 43 |
| Lymphocytes/μl | 1722 ± 93 | 1289 ± 97 | 1265 ± 142 | 1220 ± 143 | 1345 ± 286 | 1241 ± 173 | 949 ± 211 |
The number of circulating white blood cells was determined from cardiac blood in mice treated with vehicle or IL-1β at the onset of MCAo. WBC, White blood cell.
*p < 0.05 versus vehicle, Student's t test. n = 6–8 per group.
aVehicle and IL-1β groups pooled.
Marked accumulation of neutrophils in the brain was observed in vehicle- and IL-1β-treated mice at all time points assessed after ischemia. Intravascular and marginating (Fig. 5B) and parenchymal (Fig. 5C) neutrophils were observed consistently in the striatum and cerebral cortex of the occluded hemisphere. Neutrophils were more abundant and more extensively distributed in IL-1β-treated mice (Fig. 5E,G) compared with vehicle-treated mice (Fig. 5D,F), particularly in the cortex 8 h (Fig. 5D,E) and 24 h (Fig. 5F,G) after MCAo. Neutrophils were rarely observed after MCAo in the contralateral hemisphere and sham-operated mice displayed few, if any, neutrophils.
Quantification of neutrophil accumulation in the striatum revealed a general decline in numbers as reperfusion progressed but no significant differences between vehicle- and IL-1β-treated mice (Fig. 5H). In contrast, neutrophil accumulation in the cortex increased as reperfusion progressed in both vehicle- and IL-1β-treated mice. IL-1β caused a significant increase in the number of neutrophils at all time points (4 h, p < 0.05; 8 h, p < 0.05; 24 h, p < 0.01) (Fig. 5H) and resulted in an intense accumulation at 24 h.
Systemic IL-1β potentiates postischemic plasma CXC chemokine levels
We next assessed the impact of systemic IL-1β on circulating levels of CXCL1 (KC) and CXCL2 (MIP-2), the major chemokines that mobilize and direct migration of neutrophils in mice. In vehicle-treated mice, MCAo induced a fivefold increase in plasma KC concentration 4 h after MCAo (compared with sham). IL-1β induced a significant 10-fold potentiation of plasma KC concentration 4 h after MCAo (p < 0.01) (Fig. 6A). There was also a significant IL-1β-induced potentiation of plasma KC 8 h after MCAo (p < 0.05), although levels were reduced markedly compared with 4 h. The magnitude of the IL-1β-induced potentiation of plasma KC after MCAo indicated a synergistic interaction between the IL-1β and ischemic challenges. Plasma MIP-2 concentration was also elevated 4 h after MCAo and IL-1β significantly potentiated MIP-2 levels at this time point (p < 0.01) (Fig. 6B). Similarly to KC, there was a synergistic interaction between IL-1β and ischemic challenges on plasma MIP-2 concentration 4 h after MCAo. At later time points, MIP-2 concentration markedly subsided.
Figure 6.
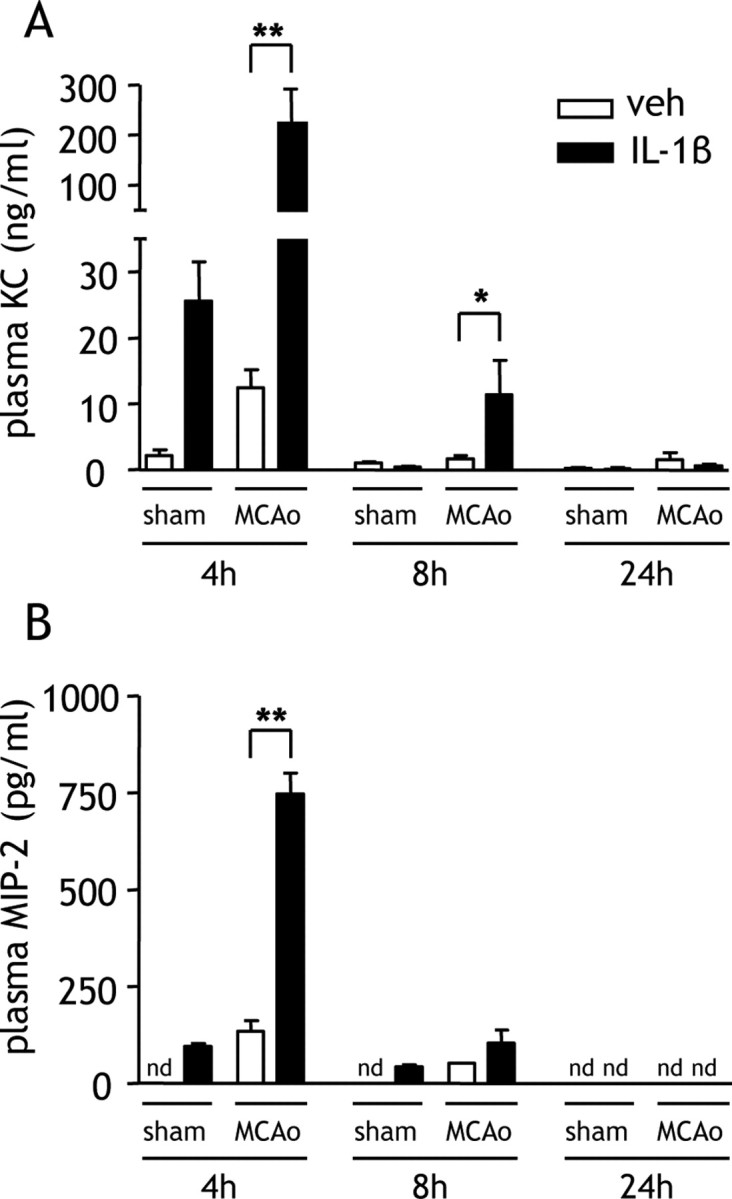
Systemic IL-1β potentiates circulating CXC chemokine levels after MCAo. Plasma KC and MIP-2 concentrations were measured by ELISA in mice administered vehicle (veh) or IL-1β systemically at the onset of MCAo. A, IL-1β significantly increased the concentration of plasma KC 4 and 8 h after MCAo, although levels were generally lower than 8 h after MCAo. By 24 h after MCAo, KC concentrations had returned to baseline levels. B, IL-1β also significantly increased the concentration of plasma MIP-2 4 h after MCAo but did not alter levels at any other time point. nd, Below detection limit. *p < 0.05, **p < 0.01 versus vehicle, Student's t test. Data are presented as mean ± SEM. n = 6–8 per group.
Neutrophils mediate the exacerbation of ischemic brain damage by systemic IL-1β
To directly test the hypothesis that neutrophils are important cellular mediators of the pathological effects of a systemic IL-1β challenge, we depleted neutrophils using an anti-PMN leukocyte-depleting antibody. The extent of neutrophil depletion was >90% (p < 0.001), and the depleting antibody had no effect on other leukocyte populations (supplemental figure, available at www.jneurosci.org as supplemental material). The number of circulating PMNs was significantly reduced by ∼90% after MCAo in PMN-depleted mice treated with vehicle (p < 0.001) or IL-1β (p < 0.001) (Fig. 7A). Abundant neutrophil accumulation in the cerebral cortex was observed after MCAo in response to IL-1β treatment in nondepleted mice (Fig. 7B). In contrast, there were few neutrophils observed after MCAo in response to IL-1β treatment in PMN-depleted mice (Fig. 7C). Quantification of neutrophil numbers in the cerebral cortex demonstrated a significant attenuation of the IL-1β-induced neutrophil infiltrate in PMN-depleted mice (p < 0.05) (Fig. 7D).
Figure 7.
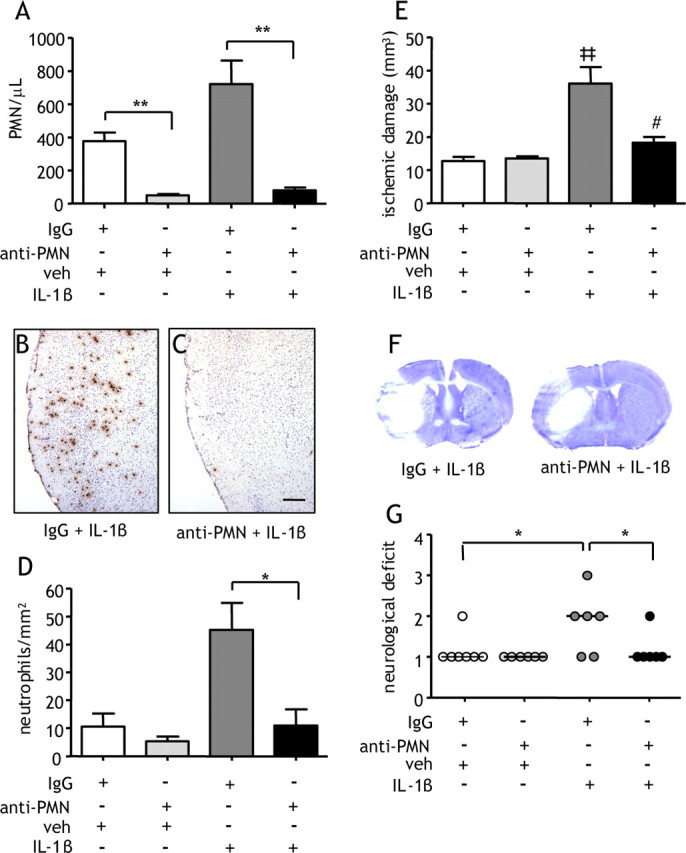
Neutropenia attenuates IL-1β-induced exacerbation of ischemic damage. Neutropenia was induced by anti-PMN antibody treatment before induction of MCAo. Mice were administered vehicle (veh) or IL-1β systemically at the onset of MCAo. A, Neutrophil depletion significantly abolished the IL-1β-induced neutrophilia 24 h after MCAo and also markedly reduced the number of circulating PMNs in control IgG-treated mice. MBS-1 immunohistochemistry shows abundant neutrophil accumulation in the cerebral cortex 24 h after MCAo in response to IL-1β treatment in control IgG-treated mice (B) but not in neutropenic mice (C). D, Quantification of MBS-1 immunohistochemistry demonstrated a significant attenuation of the IL-1β-induced neutrophil infiltrate in PMN-depleted mice. E, IL-1β induced a significant threefold increase in ischemic damage. This increase was significantly attenuated by >70% in neutropenic mice. *p < 0.05, **p < 0.01, Student's t test; ‡‡p < 0.01 versus IgG plus vehicle, #p < 0.05 versus IgG plus IL-1β, one-way ANOVA followed by Student's t test with Bonferroni's correction. Data are presented as mean ± SEM. F, Representative cresyl violet-stained brain sections illustrate a reduction in cortical damage. G, IL-1β significantly exacerbated neurological deficit, and PMN depletion significantly reduced the severity of neurological deficit in IL-1β-challenged mice. *p < 0.05, Kruskal–Wallis test followed by Dunn's multiple-comparison test. Bars show median. Scale bar, 250 μm. n = 6–7 per group.
The volume of ischemic damage 24 h after MCAo in PMN-depleted mice challenged with systemic IL-1β was significantly smaller (p < 0.05) compared with nondepleted mice in which there was a threefold increase in damage compared with vehicle-treated mice (Fig. 7E). This attenuation was the result of a reduction in cortical damage (Fig. 7F). PMN depletion also significantly reduced (p < 0.05) the severity of neurological deficit in IL-1β-challenged mice (Fig. 7G). Depletion did not alter ischemic damage or neurological deficit in vehicle-treated mice.
Discussion
We demonstrate, for the first time, that a systemic inflammatory stimulus, which mimics aspects of infection, adversely affects outcome after experimental stroke and highlight a critical role for IL-1 in mediating this effect on brain injury. Our data further implicate IL-1-induced CXC chemokines and neutrophil mobilization in this paradigm. Environmental factors, such as infection, are increasingly recognized as important modulators of the incidence of stroke and functional recovery (Emsley and Tyrrell, 2002). Infection may also interact with conventional stroke risk factors, such as atherosclerosis, to cooperatively modify stroke risk and outcome. Our data suggest that systemic inflammatory stimuli may act synergistically with the ischemic challenge to markedly amplify systemic innate immune responses leading to exacerbation of brain damage. In this way, systemic inflammatory pathways could provide a point of convergence and synergy when stroke is coincident with an attendant inflammatory stimulus.
The molecular and cellular pathways that mediate the detrimental effects of systemic inflammation on stroke are unclear. Although LPS induces the synthesis and release of multiple inflammatory mediators via TLR activation, the present data strongly implicate a critical and nonredundant function for IL-1 because coadministration of IL-1RA significantly attenuated the LPS-induced worsening of ischemic damage and neurological deficit. Consistent with this effect, systemic administration of recombinant IL-1β exacerbated ischemic damage and neurological deficit to a similar extent as LPS, suggesting that peripheral IL-1 challenge is a similar stimulus to LPS and further supporting that IL-1 is the key mediator of LPS in this brain injury paradigm. These results parallel previous studies showing that IL-1 is important for many host defense responses to systemic LPS challenge (Long et al., 1990; Luheshi et al., 1996; Miller et al., 1997b). It is likely that IL-1β was acting peripherally because levels were minimal in the circulation and undetectable in the brain after intraperitoneal administration (data not shown). In support of this, studies have shown that paracrine actions of IL-1β in peripheral tissues, such as the induction of IL-6, mediate LPS-induced fever (Miller et al., 1997a; Cartmell et al., 2000). However, this would not exclude that peripheral IL-1β may also influence the formation of damage by inducing CNS expression of cytokines and other inflammatory mediators via circulatory or neural (vagal) routes. IL-1β is a known pyrogen under nonischemic conditions, and elevated core body and brain temperature are associated with poorer outcome after stroke (Busto et al., 1987; Meden et al., 1994; Reith et al., 1996). However, previous studies have shown that the neuropathological effects of IL-1β are dissociated from its pyrogenic actions (Loddick and Rothwell, 1996; Stroemer and Rothwell, 1998). This may be explained by the compromised perfusion and metabolic activity in ischemic tissue that will reduce the influx and production of heat and mitigate the pyrogenic effects of IL-1. Indeed, using a similar rat model of stroke and magnetic resonance spectroscopy to measure brain temperature, we recently found that systemic IL-1β challenge actually reduced local brain temperature in ischemic tissue during occlusion and early reperfusion (Parry-Jones et al., 2006).
Our data provide comprehensive evidence that a systemic IL-1β challenge worsens acute brain injury by exacerbating the mobilization and infiltration of neutrophils into the ischemic brain. Previous studies in the absence of a systemic inflammatory stimulus, in which neutrophils have been depleted or their emigration from the vasculature inhibited, have provided considerable evidence that neutrophils contribute to ischemic brain damage (Chen et al., 1994; Connolly et al., 1996; Dawson et al., 1996; Yenari et al., 1998; Beray-Berthat et al., 2003; Arumugam et al., 2004), although there are also conflicting reports (Fassbender et al., 2002; Maier et al., 2004). In the present study, IL-1β potentiated the postischemic neutrophilia and accumulation of neutrophils in the cerebral cortex before alterations in brain damage. Such a temporal and spatial pattern is consistent with a causative role in the development of enhanced cortical injury and this was supported by the marked attenuation of IL-1β-induced cortical damage by neutrophil depletion. Indeed, the contribution of neutrophils to brain damage appears to be brain region specific with a predominant role in the cortex (Yenari et al., 1998; Beray-Berthat et al., 2003). This may reflect the more extensive vasculature and greater perfusion of cortical tissue (cf. striatum) that will promote neutrophil trafficking through this territory. Alternatively, more abundant cortical expression of mediators required for neutrophil extravasation (selectins, chemokines, adhesion molecules) could determine the relative predominance of cortical neutrophil accumulation. Neutrophils may promote the conversion of vulnerable yet viable cortical tissue to infarction through microvessel obstruction/thrombosis that compromises reperfusion (del Zoppo et al., 1991; Ritter et al., 2000), production of reactive oxygen species (ROS) causing oxidative stress (Aizawa et al., 2006), and release of matrix metalloproteinases that degrade components of the vascular basement membrane and extracellular matrix (Asahi et al., 2001; Gidday et al., 2005). This may account for the markedly greater BBB permeability and aggravated cerebral edema we observed in response to IL-1β challenge. These alterations have important clinical implications, because raised intracranial pressure is a precarious consequence of brain edema and a key determinant of outcome after ischemic brain injury (Ayata and Ropper, 2002). It will be important to further elucidate how the various aspects of neutrophil dynamics (e.g., mobilization, adhesion/transmigration, release of toxic mediators) contribute to the deleterious effects of systemic inflammation, for example, by specifically inhibiting endothelial–neutrophil interactions or preventing release of/inhibiting neutrophil-derived proteases and ROS. Neutrophil and total leukocyte counts were elevated in stroke patients presenting with antecedent infection (Emsley et al., 2003). Although it was not possible to assess the contribution of IL-1 to the aggravated neutrophilia in these patients, a recent phase II clinical trial in stroke patients demonstrated that IL-1RA almost completely abolished the stroke-induced neutrophilia (Emsley et al., 2005). These clinical findings lend support to the possibility that IL-1β-induced potentiation of neutrophil mobilization contributes to poorer outcome in stroke patients with preexisting systemic inflammation.
Neutrophils are only weakly responsive to IL-1β, suggesting IL-1β stimulates neutrophil mobilization indirectly. The activation and migration of neutrophils are regulated by CXC chemokines, small peptides that bind G-protein-coupled receptors on the neutrophil surface and stimulate their discharge from bone marrow and direct their local movement to sites of injury and/or infection (Baggiolini, 1998; Campbell et al., 2005). In the present study, the striking potentiation of postischemic KC and MIP-2 levels may account for the aggravated neutrophilia and cortical neutrophil accumulation induced by IL-1β. The temporal profile of the chemokine response was notable for its rapidity and transience after MCAo. This pattern is consistent with the concept that the acute but transient elevation in chemokine levels initially mobilizes neutrophils from bone marrow, and then the rapidly declining levels facilitate the detection of local tissue chemokine gradients required for neutrophil extravasation.
The present data add to the emerging evidence that systemic immune and inflammatory processes contribute to the pathophysiology of stroke. Experimental stroke induces marked activation of the peripheral immune system involving elevated cytokine and chemokine levels in peripheral lymphoid organs (Offner et al., 2006). Correlations between levels of systemic inflammatory mediators, such as cytokines and acute-phase reactants, and prognosis in stroke patients have been shown (Muir et al., 1999; Smith et al., 2004; Waje-Andreassen et al., 2005; Rallidis et al., 2006). In the present study, SAA and IL-6 levels were markedly elevated early after MCAo and potentiated by systemic IL-1β challenge, indicating an aggravated systemic acute-phase response to MCAo and highlighting the clinical relevance of our data. Peripheral infections acquired after stroke are a major complication in stroke patients (Meisel et al., 2005), and alterations in systemic immune function may also contribute to this susceptibility. Depletion of T-lymphocytes induced by sympathetic nervous system activation resulted in an immunosuppressed state and increased risk of infection after experimental stroke (Prass et al., 2003). Thus, there may be a complex series of alterations in peripheral immune pathways after stroke. Potentiation of these changes by an additional stimulus, as demonstrated in the present study, may have a critical impact on acute outcome to stroke and indicates that a fuller understanding of interactions between systemic immune pathways and the injured brain may uncover novel targets for stroke therapy. In addition, although we have focused on the acute response to ischemic brain injury, additional studies are warranted to investigate the impact of underlying systemic inflammation/immune activation on postacute reparative processes in view of the pleiotropicity of many inflammatory mediators.
In summary, the present data support integral roles for IL-1 and neutrophil mobilization in mediating the deleterious effects of a systemic inflammatory challenge on acute ischemic brain damage. This mechanistic insight may help to explain the less favorable response to stroke in patients with preceding infection. Furthermore, the concept that priming of systemic inflammatory pathways leads to exacerbated brain damage may have broad implications for stroke etiology and therapy because the majority of stroke patients are likely to present with preexisting systemic inflammation caused by comorbidities such as atherosclerosis and heart disease.
Footnotes
This work was supported by the Medical Research Council, United Kingdom. We thank Adam Denes for technical advice and critical discussion of results, and Prof. V. H. Perry for the kind gift of MBS-1 antibody.
References
- Aizawa H, Makita Y, Sumitomo K, Aburakawa Y, Katayama T, Nakatani-Enomoto S, Suzuki Y, Fujiwara K, Enomoto H, Kuroda K, Kimura T, Yahara O, Koyama S, Maruyama J, Nakamura M, Hasebe N, Kikuchi K. Edaravone diminishes free radicals from circulating neutrophils in patients with ischemic brain attack. Intern Med. 2006;45:1–4. doi: 10.2169/internalmedicine.45.1491. [DOI] [PubMed] [Google Scholar]
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Salter JW, Chidlow JH, Ballantyne CM, Kevil CG, Granger DN. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am J Physiol. 2004;287:H2555–H2560. doi: 10.1152/ajpheart.00588.2004. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Ropper AH. Ischaemic brain oedema. J Clin Neurosci. 2002;9:113–124. doi: 10.1054/jocn.2001.1031. [DOI] [PubMed] [Google Scholar]
- Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Beray-Berthat V, Croci N, Plotkine M, Margaill I. Polymorphonuclear neutrophils contribute to infarction and oxidative stress in the cortex but not in the striatum after ischemia-reperfusion in rats. Brain Res. 2003;987:32–38. doi: 10.1016/s0006-8993(03)03224-4. [DOI] [PubMed] [Google Scholar]
- Busto R, Dietrich WD, Globus MY, Valdes I, Scheinberg P, Ginsberg MD. Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. 1987;7:729–738. doi: 10.1038/jcbfm.1987.127. [DOI] [PubMed] [Google Scholar]
- Calkins CM, Bensard DD, Shames BD, Pulido EJ, Abraham E, Fernandez N, Meng X, Dinarello CA, McIntyre RC., Jr IL-1 regulates in vivo C-X-C chemokine induction and neutrophil sequestration following endotoxemia. J Endotoxin Res. 2002;8:59–67. [PubMed] [Google Scholar]
- Campbell SJ, Perry VH, Pitossi FJ, Butchart AG, Chertoff M, Waters S, Dempster R, Anthony DC. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am J Pathol. 2005;166:1487–1497. doi: 10.1016/S0002-9440(10)62365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell T, Poole S, Turnbull AV, Rothwell NJ, Luheshi GN. Circulating interleukin-6 mediates the febrile response to localised inflammation in rats. J Physiol (Lond) 2000;526:653–661. doi: 10.1111/j.1469-7793.2000.00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chopp M, Zhang RL, Bodzin G, Chen Q, Rusche JR, Todd RF., III Anti-CD11b monoclonal antibody reduces ischemic cell damage after transient focal cerebral ischemia in rat. Ann Neurol. 1994;35:458–463. doi: 10.1002/ana.410350414. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Jr, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, Stern DM, Solomon RA, Gutierrez-Ramos JC, Pinsky DJ. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97:209–216. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Ruetzler CA, Carlos TM, Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and microcirculatory perfusion in acute stroke in the SHR. Keio J Med. 1996;45:248–252. doi: 10.2302/kjm.45.248. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Schmid-Schonbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22:1276–1283. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Tyrrell PJ. Inflammation and infection in clinical stroke. J Cereb Blood Flow Metab. 2002;22:1399–1419. doi: 10.1097/01.WCB.0000037880.62590.28. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Smith CJ, Gavin CM, Georgiou RF, Vail A, Barberan EM, Hallenbeck JM, del Zoppo GJ, Rothwell NJ, Tyrrell PJ, Hopkins SJ. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J Neuroimmunol. 2003;139:93–101. doi: 10.1016/s0165-5728(03)00134-6. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K, Ragoschke A, Kuhl S, Szabo K, Fatar M, Back W, Bertsch T, Kreisel S, Hennerici M. Inflammatory leukocyte infiltration in focal cerebral ischemia: unrelated to infarct size. Cerebrovasc Dis. 2002;13:198–203. doi: 10.1159/000047776. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol. 2005;289:H558–H568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Grau AJ, Buggle F, Heindl S, Steichen-Wiehn C, Banerjee T, Maiwald M, Rohlfs M, Suhr H, Fiehn W, Becher H. Recent infection as a risk factor for cerebrovascular ischemia. Stroke. 1995a;26:373–379. doi: 10.1161/01.str.26.3.373. [DOI] [PubMed] [Google Scholar]
- Grau AJ, Buggle F, Steichen-Wiehn C, Heindl S, Banerjee T, Seitz R, Winter R, Forsting M, Werle E, Bode C. Clinical and biochemical analysis in infection-associated stroke. Stroke. 1995b;26:1520–1526. doi: 10.1161/01.str.26.9.1520. [DOI] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. 1996;16:932–940. doi: 10.1097/00004647-199609000-00017. [DOI] [PubMed] [Google Scholar]
- Long NC, Otterness I, Kunkel SL, Vander AJ, Kluger MJ. Roles of interleukin 1 beta and tumor necrosis factor in lipopolysaccharide fever in rats. Am J Physiol. 1990;259:R724–R728. doi: 10.1152/ajpregu.1990.259.4.R724. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Luheshi G, Miller AJ, Brouwer S, Dascombe MJ, Rothwell NJ, Hopkins SJ. Interleukin-1 receptor antagonist inhibits endotoxin fever and systemic interleukin-6 induction in the rat. Am J Physiol. 1996;270:E91–E95. doi: 10.1152/ajpendo.1996.270.1.E91. [DOI] [PubMed] [Google Scholar]
- Luheshi GN, Stefferl A, Turnbull AV, Dascombe MJ, Brouwer S, Hopkins SJ, Rothwell NJ. Febrile response to tissue inflammation involves both peripheral and brain IL-1 and TNF-alpha in the rat. Am J Physiol. 1997;272:R862–R868. doi: 10.1152/ajpregu.1997.272.3.R862. [DOI] [PubMed] [Google Scholar]
- Maier CM, Hsieh L, Yu F, Bracci P, Chan PH. Matrix metalloproteinase-9 and myeloperoxidase expression: quantitative analysis by antigen immunohistochemistry in a model of transient focal cerebral ischemia. Stroke. 2004;35:1169–1174. doi: 10.1161/01.STR.0000125861.55804.f2. [DOI] [PubMed] [Google Scholar]
- Meden P, Overgaard K, Pedersen H, Boysen G. The influence of body temperature on infarct volume and thrombolytic therapy in a rat embolic stroke model. Brain Res. 1994;647:131–138. doi: 10.1016/0006-8993(94)91407-9. [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Miller AJ, Luheshi GN, Rothwell NJ, Hopkins SJ. Local cytokine induction by LPS in the rat air pouch and its relationship to the febrile response. Am J Physiol. 1997a;272:R857–R861. doi: 10.1152/ajpregu.1997.272.3.R857. [DOI] [PubMed] [Google Scholar]
- Miller AJ, Hopkins SJ, Luheshi GN. Sites of action of IL-1 in the development of fever and cytokine responses to tissue inflammation in the rat. Br J Pharmacol. 1997b;120:1274–1279. doi: 10.1038/sj.bjp.0701049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir KW, Weir CJ, Alwan W, Squire IB, Lees KR. C-reactive protein and outcome after ischemic stroke. Stroke. 1999;30:981–985. doi: 10.1161/01.str.30.5.981. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Palasik W, Fiszer U, Lechowicz W, Czartoryska B, Krzesiewicz M, Lugowska A. Assessment of relations between clinical outcome of ischemic stroke and activity of inflammatory processes in the acute phase based on examination of selected parameters. Eur Neurol. 2005;53:188–193. doi: 10.1159/000086355. [DOI] [PubMed] [Google Scholar]
- Parry-Jones AR, Liimatainen T, Rothwell NJ, Kauppinen RA, Grohn OH. The effect of Interleukin-1 on local brain temperature during focal cerebral ischemia in the rat; A 1H magnetic resonance spectroscopic imaging study. Proc Int Soc Magn Reson Med. 2006;14:1452. [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallidis LS, Vikelis M, Panagiotakos DB, Rizos I, Zolindaki MG, Kaliva K, Kremastinos DT. Inflammatory markers and in-hospital mortality in acute ischaemic stroke. Atherosclerosis. 2006;189:193–197. doi: 10.1016/j.atherosclerosis.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Reith J, Jorgensen HS, Pedersen PM, Nakayama H, Raaschou HO, Jeppesen LL, Olsen TS. Body temperature in acute stroke: relation to stroke severity, infarct size, mortality, and outcome. Lancet. 1996;347:422–425. doi: 10.1016/s0140-6736(96)90008-2. [DOI] [PubMed] [Google Scholar]
- Ritter LS, Orozco JA, Coull BM, McDonagh PF, Rosenblum WI. Leukocyte accumulation and hemodynamic changes in the cerebral microcirculation during early reperfusion after stroke. Stroke. 2000;31:1153–1161. doi: 10.1161/01.str.31.5.1153. [DOI] [PubMed] [Google Scholar]
- Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. 2004;351:2611–2618. doi: 10.1056/NEJMoa041747. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Emsley HC, Gavin CM, Georgiou RF, Vail A, Barberan EM, del Zoppo GJ, Hallenbeck JM, Rothwell NJ, Hopkins SJ, Tyrrell PJ. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004;4:2. doi: 10.1186/1471-2377-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroemer RP, Rothwell NJ. Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1beta in the rat. J Cereb Blood Flow Metab. 1998;18:833–839. doi: 10.1097/00004647-199808000-00003. [DOI] [PubMed] [Google Scholar]
- Waje-Andreassen U, Krakenes J, Ulvestad E, Thomassen L, Myhr KM, Aarseth J, Vedeler CA. IL-6: an early marker for outcome in acute ischemic stroke. Acta Neurol Scand. 2005;111:360–365. doi: 10.1111/j.1600-0404.2005.00416.x. [DOI] [PubMed] [Google Scholar]
- Yenari MA, Kunis D, Sun GH, Onley D, Watson L, Turner S, Whitaker S, Steinberg GK. Hu23F2G, an antibody recognizing the leukocyte CD11/CD18 integrin, reduces injury in a rabbit model of transient focal cerebral ischemia. Exp Neurol. 1998;153:223–233. doi: 10.1006/exnr.1998.6876. [DOI] [PubMed] [Google Scholar]