




Abstract
To analyze relationships between replication and homologous recombination in mammalian cells, we used replication inhibitors to treat mouse and hamster cell lines containing tandem repeat recombination substrates. In the first step, few double-strand breaks (DSBs) are produced, recombination is slightly increased, but cell lines defective in non-homologous end-joining (NHEJ) affected in ku86 (xrs6) or xrcc4 (XR-1) genes show enhanced sensitivity to replication inhibitors. In the second step, replication inhibition leads to coordinated kinetics of DSB accumulation, Rad51 foci formation and RAD51-dependent gene conversion stimulation. In xrs6 as well as XR-1 cell lines, Rad51 foci accumulate more rapidly compared with their respective controls. We propose that replication inhibition produces DSBs, which are first processed by the NHEJ; then, following DSB accumulation, RAD51 recombination can act.
Keywords: double-strand breaks/homologous recombination/non-homologous recombination/Rad51/replication inhibition
Introduction
Faithful genome transmission requires the association of several mechanisms including replication and recombination. Replication forks are routinely arrested by a broad variety of stresses (for review see Hyrien, 2000; Rothstein et al., 2000). In bacteria, homologous recombination is an efficient process helping to reactivate a replication fork that has been blocked (for review see Kuzminov, 1995; Cox et al., 2000). Replication blocks have also been shown to induce DNA double-stranded breaks (DSBs) in recBC mutants (Bierne et al., 1997; Seigneur et al., 1998). In yeast, DNA polymerase mutants accumulate Holliday junctions at the rDNA locus, a molecular intermediate of homologous recombination (Zou and Rothstein, 1997). Moreover, repair of lesions during replication often involves recombination between sister chromatids (Fabre et al., 1984; Kadyk and Hartwell, 1992). Replication fork pausing occurs from bacteria to humans, but very little is known of the relationships between replication and recombination in mammalian cells (Cox et al., 2000; Hyrien, 2000; Rothstein et al., 2000). This link may be of crucial importance for genome stability and has been discussed in the case of cancer predisposition (Scully et al., 2000). However, the mechanisms and molecular characteristics of the connection between replication and recombination have yet to be elucidated in mammalian cells.
In order to study the precise impact of replication arrest on recombination, we used replication inhibitors to treat mammalian cell lines carrying intrachromosomal tandem repeat recombination substrates. We used a set of drugs that are well characterized and commonly used in cell synchronization experiments. These drugs specifically inhibit either the initiation (mimosine, ciclopirox olamine) or the elongation (hydroxyurea, aphidicolin) of replication (Levenson and Hamlin, 1993). Moreover, these replication inhibitors have been shown to induce DNA single-strand breaks and sister chromatid exchange (SCE) (Ishii and Bender, 1980; Fram and Kufe, 1982; Caligo et al., 1988). Different pathways can promote SCE. In the transformed chicken DT40 B-cell line, RAD51 is involved in spontaneous SCE (Sonoda et al., 1999), whereas in mammalian cells, mRAD51 and the mouse RAD54 gene do not affect spontaneous SCE and partially control induced SCE (Dronkert et al., 2000; S.Lambert and B.S.Lopez, submitted). This suggests that alternative pathways should exist. For instance, it has been suggested that topoisomerases are involved in SCE formation in mammalian cells (Dillehay et al., 1989). Thus, the effects of replication inhibition on homologous recombination and the pathways involved have yet to be clearly elucidated in mammalian cells.
In the present work, we measured and characterized molecular and genetic recombination events occurring in response to replication inhibition in mammalian cells.
Results
Replication inhibitors stall cell cycle progression in late G1 and in S phases
Cells were treated with different drugs known to inhibit replication reversibly in mammalian cells. Two of them (mimosine and ciclopirox olamine) inhibit the initiation step of the replicons and two others (aphidicolin and hydroxyurea) inhibit elongation of the replicons (Levenson and Hamlin, 1993).
The consequences for the cell cycle after different times of contact with two different concentrations of aphidicolin are shown in Figure 1A. Similar results were obtained with mouse L-cells as well as with hamster CHO cells (data not shown). After 6 h of contact with the drug, the peak of cells in the G2 phase completely disappeared whilst a G1-like peak and the S plateau were still present. This result shows that cells in the G2 phase proceed through mitosis to reach the G1 phase, but that cells in the S phase are blocked and can not reach the G2 phase. This is consistent with the inhibition of replication leading to the arrest of the cell cycle in late G1, at the entry to the S phase, and in the S phase.

Fig. 1. Arrest of cell cycle progression by replication inhibitors, measured by flow cytometry analysis. The cell cycle phases are determined by the DNA content measured by flow cytometry [5-bromo-2-deoxyuridine (BrdU) incorporation being inhibited by the drug]. (A) Effect of different times of contact with two different concentrations of aphidicolin. The aphidicolin concentrations are indicated on the left and the different times of contact are indicated at the top of the figure. (B) Cell cycle distribution after 24 h of treatment with the different drugs. Concentrations were: 200 µM mimosine; 1 mM hydroxyurea; 20 µM ciclopirox olamine; 6 µM aphidicolin.
In the present work we used a set of different replication inhibitors. We checked the cell cycle profiles after 24 h of contact with the different drugs (Figure 1B). The disappearance of G2-phase cells was observed with all the drugs, and is consistent with a block in late G1 and S phases with all the replication inhibitors.
Thus, as expected, treatment with the different replication inhibitors blocked the progression in the S phase. Taking into account the DNA content, cells arrested in the S phase should have accumulated stalled replication intermediates. This strategy allowed us to address the question of whether the stalling of the replication forks actually results in DNA breaks and in stimulation of homologous recombination in mammalian cells.
Replication inhibitors induce DNA breaks
Arrest of replication fork progression should lead to the accumulation of single-strand nicks resulting from unsealed Okasaki fragments, or single-strand gaps between two replicons. Additionally, these nicks and single-strand gaps could also be processed into double-stranded breaks. First, we measured the induction of DNA breaks using the COMET assay under alkaline conditions, which allow detection of both single- and double-stranded breaks, in addition to alkali labile sites. The distribution of the classes of comet tail moments after treatment with either hydroxyurea or mimosine is shown in Figure 2A. Both hydroxyurea and mimosine induced DNA breaks, but hydroxyurea was much more efficient (Figure 2A). Other drugs such as aphidicolin also led to the accumulation of breaks in the DNA (data not shown).
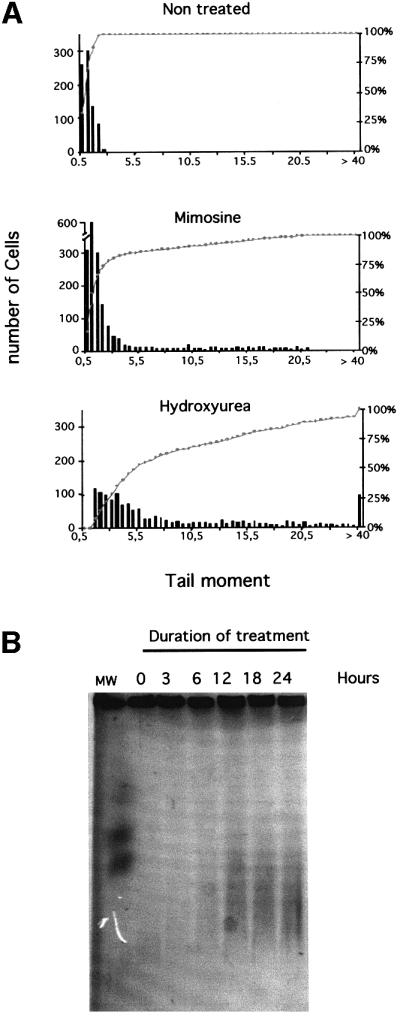
Fig. 2. DNA breaks induced by replication inhibitors. (A) The COMET assay measures single- and double-strand breaks. The amount of breaks is estimated by the tail moment: fraction of the DNA in the tail × distance between the mass center of the head and the mass center of the tail. The histograms correspond to the tail moments after 24 h of treatment with the replication inhibitors (indicated on the tops of the histograms). The bars correspond to the number of cells with the value of tail moment. The amount of breaks is correlated with the tail moment values. The curves (gray squares) indicate the cumulative percentage of cells. (B) PFGE detects the DSBs. The different times of contact with hydroxyurea are indicated on the top of the gel.
DSBs induced by hydroxyurea treatment were specifically analyzed by pulsed-field gel electrophoresis (PFGE) (Figure 2B). The smears indicate an accumulation of DSBs as a function of the time of replication block. DSB smears were slightly detectable after 6 h of blockage and were then clearly visible for 12, 18 and 24 h of blockage. However, it is possible that low and undetectable amounts of DSB were produced during the 3 h treatment. Nevertheless, this result indicates that increasing the time of replication block leads to an accumulation of DSB.
Recombination is stimulated by replication elongation inhibitors in replicating S phase cells
We compared the efficiency of replication initiation inhibitors and elongation inhibitors for the stimulation of recombination. Recombination was measured in the CHO-DRA10 line (Liang et al., 1998) using the strategy depicted in Figure 3A. Two different inhibitors of replicon initiation (mimosine and ciclopirox olamine) moderately induced homologous recombination. In contrast, two different replicon elongation inhibitors (aphidicolin and hydroxyurea) strongly stimulated homologous recombination (Figure 3B). In the CHO-DRA10 line, inhibition of replication elongation stimulates recombination 15- to 25-fold more efficiently than inhibition of replication initiation.

Fig. 3. Induction of recombination by elongation inhibitors in S phase. (A) The cell line used is hamster CHO-DRA10 containing the recombination substrate drawn and described elsewhere (Liang et al., 1998). This substrate contains a direct repeat of two inactive neomycin-resistant (NEO) genes; parental cells are sensitive to G418. Recombination restores a functional NEO gene; recombinants are resistant to G418. The frequency of spontaneous recombination is 6 × 10–7. (B) Induced recombinant corresponds to the number of G418-resistant clones for 107 viable treated cells subtracted from the number of G418-resistant clones in 107 non-treated viable cells. Cells were treated for 24 h with the drugs or the combinations indicated below the bars. Concentrations were: 200 µM mimosine; 1 mM hydroxyurea; 20 µM ciclopirox olamine; 0.6 µM aphidicolin. (C) Synchronization (indicated as ‘synchro’ in the figure) by double thymidine block. The DNA content is measured by FACS at different times after the release of the second thymidine block. H+0h: 0 h after the thymidine block release; H+1h: 1 h after the thymidine block release; H+2h: 2 h after the thymidine block release; H+3h: 3 h after the thymidine block release. The percentage of cells in the S phase is indicated on the histogram. (D) Induced recombinant under the different conditions. Synchro: synchronization by double thymidine block; HU: treatment with hydroxyurea for 24 h. H+ 0, 1, 2 or 3 h indicates the moment of hydroxyurea addition after the thymidine block release. (E) HU-induced recombinant as a function of the percentage of cells in the S phase at the beginning of the HU treatment. These values correspond to the progression in S phase (see Figure 3C).
The inhibitors used here are generally considered to be highly specific for replication inhibition. Nevertheless, we verified whether they act specifically on replicating cells in the S phase. We measured whether they actually act on a common pathway for stimulation of recombination, by treating the cells with a combination of two inhibitors. If they act on two different pathways, the combination of two drugs should stimulate recombination more efficiently than each drug alone. In contrast, if the two drugs act on the same pathway, the two-drugs combination should stimulate recombination as efficiently as one drug alone, in a kind of ‘pharmacological epistasis’. Figure 3B shows that hydroxyurea and aphidicolin in tandem stimulated recombination as efficiently as hydroxyurea or aphidicolin alone. Thus, they stimulate recombination by acting on a common pathway, i.e. the inhibition of replication elongation. Similarly the ciclopirox olamine/mimosine combination stimulated recombination to the same extent as ciclopirox olamine or mimosine alone. Finally, the combination of an initiation inhibitor (mimosine) with one elongation inhibitor (hydroxyurea or aphidicolin), on non-synchronized cells, stimulated recombination as efficiently as hydroxyurea or aphidicolin alone, i.e. the most efficient inductor (Figure 3B). These results show that the drugs stimulate recombination by a pathway common to all of them; this pathway must be the inhibition of replication.
If the recombination stimulation is correlated with the block of the replication, it should specifically act on S phase cells. To verify this interpretation, we synchronized the hamster CHO-DRA10 line cells before treating them with hydroxyurea.
Synchronization by double thymidine block is shown in Figure 3C. An asynchronous population contains 30–40% of cells in the S phase. Synchronization resulted in 70–90% of cells in the S phase depending on the time after the thymidine block release, i.e. a 2- to 3-fold increase in the number of S phase cells (Figure 3D).
Double thymidine block treatment only poorly induced recombination compared with treatment with hydroxyurea (Figure 3D). Hydroxyurea was then added at different times after the release of the thymidine block and maintained for 24 h. Hydroxyurea induced recombination 2–3 times more efficiently in cells synchronized in the S phase compared with non-synchronized cells. This factor corresponds to the increase in the frequency of cells in the S phase in a synchronized population (compare Figure 3C and D). Remarkably, the extent of recombination stimulation was linearly correlated with the percentage of cells in the S phase, i.e. the time between the block release and the hydroxyurea treatment (Figure 3E). These results suggest that the homologous recombination was more efficient during the progression in the S phase.
Taken together, the results show that the drugs stimulate homologous recombination via the inhibition of replication (mainly elongation) in mammalian cells.
Stimulation of homologous recombination by aphidicolin as a function of the time of blockage
We then analyzed the recombination stimulation at different times of contact with aphidicolin, i.e. different times of replication inhibition. We first measured recombination induced by replication inhibitors (RIRI) in mouse L-cells (i) to verify the phenomenon in another cell line type and (ii) because a couple of mouse L-cells provided the opportunity to measure the RIRI with direct or inverted repeat recombination substrates (Figure 4A). Direct repeats and inverted repeats do not monitor exactly the same recombination pathways (for review see Klein, 1995): direct repeats monitor gene conversion as well as single-strand annealing (SSA) events. Inverted repeats can not measure SSA but mainly monitor gene conversion plus non-SSA alternative pathways. As observed for the cell cycle arrest, the two different concentrations of aphidicolin stimulate recombination in a similar range (Figure 4B and C). Aphidicolin stimulated recombination as a function of the duration of the treatment, i.e. of replication inhibition. But two phases of stimulation were observable: the recombination increase was linear but modest for times of contact between 6 and 18 h, even though (i) fluorescence-activated cell sorting (FACS) analysis indicated a replication block for these times of treatment and (ii) PFGE showed the appearance of DSB at 6 h. Recombination was then strongly stimulated for times of contact with aphidicolin comprised between 18 and 24 h (Figure 4B). Similar results were obtained with direct repeats (Figure 4B) and with inverted repeat recombination substrates (Figure 4C). Interestingly, the kinetic of the recombination process per se was correlated with the kinetics of DSB accumulation (compare Figures 2B and 4). Moreover, comparison of these two kinetics highlights the fact that a huge amount of DSBs is required to stimulate homologous recombination efficiently. This may reflect the probability of introducing one DSB in the recombination substrate. Similar kinetics were observed with the hamster CHO-DRA10 line treated with hydroxyurea (see below, Figure 5C).

Fig. 4. Treatment with aphidicolin induces recombination as a function of the time of contact with the drug. (A) The cell lines derive from mouse L-cells. The recombination substrates are depicted on the figure and have been described elsewhere (Liskay et al., 1984). The cell line pJS3-10 contains direct repeat recombination substrates (on the left). The cell line pJS4-7-1 contains inverted repeat recombination substrates (on the right). (B) Induction of recombination between direct repeats (pJS3-10, on the left) or between inverted repeats (pJS4-7-1, on the right). The duration of treatment and the aphidicolin concentrations are indicated in the figure. (C) Example of Southern blotting of recombinant clones (probed with a TK sequence). On the left panel is the restriction map of the direct repeats substrate. After triple digestion (HindIII, BamHI, XhoI), a deletion event will produce a 6 kb (HindIII–BamHI) fragment. Gene conversion will produce either 5, 1.8 and 1 kb bands or 6, 1.3 and 0.5 kb bands; however, the 0.5 kb bands are usually hard to detect in these kinds of gels (Liskay et al., 1984). On the right is one example of Southern blotting for gene conversion events. The numbers on the top correspond to different clones double-resistant HATR(Tk+)/G418R(Neo); nr: non-recombinant parental line (pJS3-10). The sizes of the restriction fragments are indicated on the left side of the panel.

Fig. 5. Role of RAD51 in the induction of recombination by hydroxyurea. (A) and (B) Rad51 foci accumulation during hydroxyurea treatment. (A) Example of Rad51 foci. (B) Percentage of cells with Rad51 foci as a function of the time of contact (h) with hydroxyurea. The percentage of cells in the S phase for each time of contact with hydroxyurea is indicated in the figure. (C) and (D) Effect of the status of RAD51 on the RIRI. (C) Recombination induction as a function of the duration of treatment. (D) Survival (cloning efficiency) as a function of the duration of treatment. The duration of treatment is indicated in the figure. The names of the cell lines are indicated in the figure. The values correspond to the mean of three independent experiments. Error bars are omitted to avoid overloading the figure since the percentages of survival are not significantly different. CHO-DRA10 is the parental cell line; C2 is CHO-DRA10 transfected with the empty vector. These two lines are control lines. F1.4.1 and F1.4.2 are two independent lines expressing the RAD51 dominant-negative form (hypo-rec). Rm2 and Rm4 are two independent lines over-expressing MmRAD51 (hyper-rec).
Experiments with the inverted repeat substrate suggest that gene conversion (which is an important pathway with such a substrate) may arise. With direct repeats, gene conversion, without associated crossing over, should lead to a double HATR/G418R-resistant clone. Deletion events resulting from either gene conversion associated with crossing over or SSA lead to clones that are resistant to HATR but sensitive to G418 (Figure 4C). After 24 h of treatment with aphidicolin, 50% of the recombinant clones were HATR/G418R, suggesting a high proportion of gene conversion events. To demonstrate the occurrence of gene conversion events, we studied the molecular structure of double HATR/G418R-resistant recombinants by Southern blotting. Figure 4C shows an example of six double HATR/G418R clones, demonstrating the existence of the two predicted classes of gene conversion events. Thus, inhibition of replication by aphidicolin results in a high proportion (at least 50%) of gene conversion stimulation.
Role of RAD51 in RIRI
The mammalian Rad51 protein is homologous to the yeast recombination protein ScRad51 (Morita et al., 1993; Shinohara et al., 1993) and is involved in gene conversion associated with DSB repair (Lambert and Lopez, 2000). Since replication inhibitors stimulate gene conversion, we tested whether RIRI was affected by RAD51.
The Rad51 protein has been described as re-localizing in nuclear foci after a genotoxic stress (Haaf et al., 1995). These foci are generally interpreted as DNA repair foci. We analyzed the kinetic of Rad51 foci formation during treatment with hydroxyurea (Figure 5A and B). A moderate increase in foci formation was observed for 6 h of treatment, then a strong accumulation of Rad51 foci was recorded for times of contact of 12 and 18 h. These foci do not correspond to S phase foci because the frequency of cells with Rad51 foci varies greatly, even though the percentage of S phase cells is comparable in each condition. In addition, the kinetics of Rad51 foci formation are superimposable on the kinetics of DSB accumulation. Moreover, the kinetics of DSB and foci accumulation are strongly consistent with the kinetics of recombination stimulation. Taken together, these results argue for an involvement of Rad51 in the cellular response to replication inhibition, but at a late stage.
We then tested genetically the involvement of RAD51 in the RIRI and whether this pathway was essential for cell viability. We have devised CHO-DRA10 derivative lines expressing either the mouse MmRAD51 cDNA or a chimera SMRAD51 corresponding to the fusion of the 55 N-terminal amino acids from the yeast ScRAD51 to MmRAD51. MmRAD51 stimulates recombination, whereas SMRAD51 inhibits spontaneous as well as DSB-induced (γ-rays, I-SceI) recombination in CHO-DRA10 lines, without affecting cell viability (Lambert et al., 1999). Thus, we tested whether the expression of these different forms of RAD51 affects the RIRI.
The expression of the different RAD51 forms affected the two recombination phases of the RIRI differently (Figure 5C). RAD51 has no effect on the early phase but specifically affected the second phase of recombination stimulation. For replication block lower than 12 h no significant differences were observed between the lines expressing the different RAD51 forms. When replication inhibition was prolonged for longer than 12 h, recombination was strongly increased. These times of blockage correspond to the accumulation of DSB (Figure 2) and of Rad51 foci (Figure 5B). The recombination increase was 4- to 5-fold stimulated by the expression of the wild-type MmRAD51, whilst expression of the dominant-negative SMRAD51 inhibited the RIRI 6- to 8-fold, for 24 h of replication inhibition (Figure 5C). Thus, RAD51 especially affects the second phase of the cell response to replication inhibition. This second phase corresponds to the accumulation of DSBs and to the actual stimulation of recombination. However, treatment with hydroxyurea led to the same toxicity in the different lines, showing that the expression of the different RAD51 forms does not affect cellular sensitivity to replication inhibition (Figure 5D). These results also show that it is possible to decrease the efficiency of this pathway compared with control cell lines, without affecting cell viability compared with control cell lines.
NHEJ is involved in resistance to replication inhibitors in the early response
The treatment with replication inhibitors was toxic in the early cell response, but induced DSB formation and stimulated homologous recombination in the late cell response. One hypothesis is that an alternative DSB repair pathway acts in the early response. NHEJ is a good candidate for such an early pathway (Siede et al., 1996; Takata et al., 1998; Essers et al., 2000). We used two mutant cell lines affected in two different genes involved in NHEJ: xrs6 is mutated in ku86, and XR-1 is mutated in xrcc4. These mutant cell lines derive from different parental lines (CHO-K1 for xrs6 and 4364 for XR-1) and they are maintained in different media (see Materials and methods). Thus, they should be compared with their respective controls.
A significantly higher sensitivity to hydroxyurea was observed in the NHEJ-defective cell lines (both in xrs6 and in XR-1) during the early response (Figure 6A and B). Differences in sensitivity were still measurable after 6, 12 and 18 h of treatment, but increasing the time of contact with hydroxyurea reduced these differences. Ku86 protein is involved in DNA ends recognition, whereas Xrcc4 protein is a cofactor of the ligase 4 and is thus involved in a late step of NHEJ. To verify that the sensitivity to hydroxyurea, observed in the early step, corresponds to an actual defect in NHEJ, we complemented the XR-1 line with the XRCC4 cDNA. In two independent complemented cell lines (namely X4C and X4V) the expression of the XRCC4 cDNA restored the sensitivity to the level of the control 4364 line (Figure 6C).

Fig. 6. Sensitivity of the ku86– mutant xrs6 line (A) and of the xrcc4– mutant XR-1 line (B), or the XR-1 line complemented with the XRCC4 cDNA (C), as a function of the duration of treatment with hydroxyurea. The names and phenotypes of the cell lines are indicated in the figure. X4C and X4V are two independent XR-1 deriving clones, complemented with the XRCC4 cDNA.
Taken together, these results suggest that following replication inhibition, DSBs are accumulated. NHEJ would take place first, then when NHEJ became overwhelmed by DSB accumulation, homologous recombination would act efficiently in the late stage. To test this interpretation, we measured the kinetics of Rad51 foci formation after treatment with hydroxyurea in the xrs6 and the XR-1 mutant cell lines compared with their respective control cell lines and with XR-1 deriving lines complemented with XRCC4 cDNA (Figure 7). The kinetics show that the maximum of foci accumulation was reached between 9 and 12 h after the beginning of treatment in the xrs6. The plateau of Rad51 foci accumulation was delayed for several hours in the corresponding CHO-K1 control line (Figure 7A). It could be argued that the difference in the kinetics could be due not to a defect in NHEJ but to an abnormal high level of Ku86 protein in the CHO-K1 cell line, resulting in competition on the DSB and in a delay of Rad51 foci formation. Thus, we tested the kinetics of Rad51 foci formation induced by hydroxyurea in the xrcc4-defective line. Similarly to the xrs6 line, the plateau level of Rad51 foci was reached between 9 and 12 h of treatment in the XR-1 line, whereas this plateau was delayed in the wild-type 4364 control line (Figure 7B). Interestingly, in two independent clones complemented with the XRCC4 cDNA, the kinetic of Rad51 foci formation became similar to that of the wild-type 4364 control cell line (Figure 7B). Thus, the functional NHEJ delays the occurrence of Rad51 foci induced by replication inhibitors.

Fig. 7. Kinetics of Rad51 foci accumulation in the ku86– xrs6 line (A) or the xrcc4– XR-1 line and its derivatives (B). The values indicate the percentage of cells with Rad51 foci, as a function of the time of contact with hydroxyurea. At least 300 cells were counted per point. The names and phenotypes of the cell lines are indicated in the figure. X4C and X4V are two independent XR-1 deriving clones, complemented with the XRCC4 cDNA.
After treatment with hydroxyurea, a defect in NHEJ, either due to Ku86 protein inactivation or XRCC4 mutation, results in an increased sensitivity in the early steps associated with an accelerated kinetic of Rad51 foci formation.
Discussion
In the present report, we show that replication inhibitors stimulate recombination of cells in the replicating S phase. Prolonged treatments with replication inhibitors result in accumulation of DSBs. The strategy used here gave us the opportunity to compare the kinetics of DSB accumulation, Rad51 foci formation, and stimulation of the homologous recombination process per se. We observed a striking correlation between these three kinetics. In addition, these data afford additional evidence on the putative role of Rad51 foci in DSB repair.
The extent of RIRI is directly correlated with progression in S phase. This result is consistent with the expression pattern of Rad51 protein in the S and G2 phases (Yamamoto et al., 1996; Chen et al., 1997; Vispé et al., 1998). In addition, the combination of different inhibitors stimulates recombination as efficiently as treatment with only one inhibitor in a kind of ‘pharmacological epistasis’. These results show that the different inhibitors act on recombination via a common target, i.e. replication inhibition.
We used two categories of well characterized drugs known to inhibit either replicon initiation or elongation. In each category, the results are extremely homogeneous since the two drugs of each category provoke the same effect: the two elongation inhibitors strongly stimulate recombination whereas the two initiation inhibitors show moderate effects on recombination. These results are consistent with the fact that elongation inhibitors produce more DNA breaks than initiation inhibitors. The results are also consistent with the synchronization experiments showing that cells in late S phase, which should present more elongated replicons, are more proficient for the RIRI.
Replication inhibition stimulates recombination between both direct repeats and inverted repeat recombination substrates. Recombination between direct repeats uses gene conversion as well as RAD51-independent SSA pathways; inverted repeat recombination mainly uses the gene conversion pathway plus alternative non-SSA pathways (for review see Klein, 1995; Rattray and Symington, 1995). Since RIRI acts on both types of substrate, these results indicate that RIRI involves a common pathway, i.e. the gene conversion pathway. We confirm molecularly by Southern blotting the occurrence of gene conversion events. Moreover, treatment with hydroxyurea leads to Rad51 foci formation with kinetics consistent with those of DSB accumulation and recombination stimulation. Finally, over-expression of the wild-type MmRAD51 stimulates RIRI, whereas expression of the trans-dominant-negative SMRAD51 impairs RIRI. Taken together, these results demonstrate the involvement of RAD51 in RIRI. RAD51 is an essential gene in vertebrates (Tsuzuki et al., 1996; Sonoda et al., 1998). In chicken DT40 transformed cell line, it has been suggested that the essential role of RAD51 is to repair spontaneous lesion during replication by using the sister chromatid (Sonoda et al., 1999). Since RAD51 is also essential in mammalian cells, the hypothesis is commonly extrapolated from chicken to mammalian cells. Our results show that RAD51-dependent recombination is stimulated by replication arrest but in the late response to replication inhibitors. Moreover, the use of a trans-dominant-negative form of RAD51 has shown that it is possible to substantially decrease spontaneous as well as DSB-induced recombination without affecting viability of the hamster CHO cells (Lambert and Lopez, 2000). In addition, we show here that the RAD51 trans-dominant-negative form also significantly inhibits the RIRI without affecting cell viability. However, it can not be excluded that a low level of recombination could be sufficient to ensure cell viability. Nevertheless, it is possible to substantially decrease RIRI without affecting cell viability.
The time-course of replication elongation inhibition shows two steps in the cell response. The first step corresponds to the time of blockage between 0 and 12 h. During this period the toxicity is correlated with the time of contact with the drug, but homologous recombination is only moderately increased. Two different NHEJ-defective cell lines, affected in two different genes (ku86 or xrcc4), show an enhanced sensitivity to elongation inhibitors during this early step. These results suggest that NHEJ preferentially acts during this step, compared with homologous recombination. The second step corresponds to prolonged arrest (>12 h). During this step DSBs accumulate, the toxicity reaches a maximum and the sensitivity of the NHEJ-defective cells reaches that of the control line. Moreover, Rad51 foci accumulate during this phase. In addition, homologous recombination is strongly stimulated during this second step and this pathway is affected by the status of the exogenous RAD51 expressed. These results indicate that the second step corresponds to a RAD51-dependent pathway and that a huge amount of DSBs is required to stimulate homologous recombination. Remarkably, in the present experiments we observed a good correlation between the kinetics of DSB production, Rad51 foci formation and recombination. Taken together, these results suggest that NHEJ and RAD51 recombination act in a temporal sequence. One model could be proposed (Figure 8): (i) in the early phase, replication arrest leads to DSB formation. NHEJ and homologous recombination could compete for the DSBs (Haber, 1999; Van Dyck et al., 1999), but NHEJ would be much more efficient. A defect in NHEJ results in an increased sensitivity to replication inhibitors. (ii) In the late phase, DSBs are accumulated and could saturate the NHEJ machinery, resulting in DSBs accessible to the homologous recombination machinery. The fact that stimulating or inhibiting the RAD51 recombination pathway does not modify the cell sensitivity agrees with the hypothesis of NHEJ saturation. Alternatively, NHEJ and homologous recombination would not compete, but their action would be regulated and coordinated by the cell in a temporal sequence: (1) NHEJ for most of the damage to ensure cell viability, then (2) homologous recombination for the residual damage. These models are supported by the fact that in the absence of a functional NHEJ pathway (defect of either Ku86 or Xrcc4), Rad51 foci accumulate faster compared with control cell lines or XR-1 lines complemented with the XRCC4 cDNA.

Fig. 8. NHEJ and homologous recombination on DSB induced by replication inhibitors. Replication arrest creates DSBs. In the early response, DSBs are mainly processed by NHEJ and homologous recombination is marginally involved. A defect in NHEJ leads to hypersensitivity to replication inhibitors. Prolonged arrest (late response) leads to the accumulation of DSBs. NHEJ would be overwhelmed and homologous recombination can act efficiently.
The processes described here may have various important biological consequences. The control of genome integrity is essential to prevent neoplastic development. The data presented here detail the genetic and molecular connections between two essential processes of genome maintenance in mammalian cells: replication and homologous recombination.
Materials and methods
DNA manipulations
All DNA manipulations were performed as described (Sambrook et al., 1989; Ausubel et al., 1999).
Cells and Rad51 foci
CHO-DRA10, mouse pJS3-10 and pJS4-7-1 cell lines were cultured at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). xrs6 (ku86 mutant cell line) and CHO-K1 (the corresponding wild-type cell line) were cultured in α-MEM supplemented with 10% FBS. XR-1 (xrcc4 mutant cell line) and 4364 (the corresponding wild type) were cultured in DMEM without pyruvate sodium. The Rad51 foci were analyzed as described (Haaf et al., 1995) using an anti-Rad51 antibody (Oncogene research).
Measurement of recombination
Strategies and cell lines used to measure recombination. The pJS3-10 and pJS4.7.1 lines are sensitive to the HAT selective medium. Recombinant TK+ clones were selected in HAT medium (100 µM hypoxanthine, 2 µM aminopterin, 15 µM thymidine) as described (Liskay et al., 1984). CHO-DRA10 and lines Rm2, Rm4, F1-4.1, F1-4.2 derived from CHO-K1, contain a recombination substrate (Liang et al., 1998) and express different Rad51 forms (Lambert and Lopez, 2000). NEOR recombinants were selected with 500 µg/ml G418. The frequency of recombination is given by the ratio of the number of HAT-resistant clones or G418 (NEOR)-resistant clones to the total number of viable cells.
Recombination frequency after replication inhibitors. Cells were incubated at the concentration and for the time indicated, and were then trypsinized and divided into two fractions. The first fraction was used to calculate the viability by measuring the plating efficiency. The second fraction was plated under HAT or G418 selection to measure the frequency of TK+ or NEOR clones. The recombination frequency was estimated by the ratio: number of TK+ or NEOR clones to the total number of surviving clones.
COMET assay
The protocol for the alkaline COMET assay was adapted from that initially reported (Singh et al., 1988). Cells were suspended in Ca2+- and Mg2+-free phosphate-buffered saline (PBS), then mixed with low-melting-point agarose (Type VII; Sigma) maintained at 37°C, in order to obtain a final concentration of 0.5% agarose and ∼5 × 105 cells/ml. One hundred and fifty microlitres of this cell suspension were loaded on a non-frosted slide pre-coated with 0.5% air-dried agarose. After 5 min over ice, the slides were placed in freshly prepared lysis buffer for 1 h 30 min at 4°C [2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris pH 10, 1% sodium sarcosinate, 1% Triton X-100, 1% dimethylsulfoxide (DMSO)]. Slides were then transferred to a horizontal electrophoresis unit filled with fresh alkaline buffer (300 mM NaOH, 1 mM Na2EDTA pH 13) at room temperature. The slides were left for 25 min to allow the DNA to unwind and then electrophoresis was conducted for 25 min at 21 V and 300 mA. After electrophoresis, the slides were drained, rinsed twice for 5 min with 0.4 mM Tris pH 7.5 and once with double-distilled water. The slides were dried for 30 min at 40°C, dehydrated in a 100% ethanol bath, dried thoroughly at 37°C and stored at room temperature until the analysis. Before analysis, the slides were re-hydrated and stained for 30 min in a 5 µg/ml propidium iodide solution and covered with a coverslip for immediate analysis. A total of 800 cells on three slides were observed at 200× magnification using an epifluorescence microscope (LEICA DMLB) equipped with an excitation filter of 515–560 nm, a 50 W mercury lamp, and a barrier filter at 590 nm. Comets were captured and analyzed with the fully automated system Morphostar Comet (IMSTAR s.a.). Tail moment (Olive et al., 1990) was measured.
Pulsed-field gel electrophoresis
Cells were prepared for PFGE as described (Gunderson and Chu, 1991; Dhermain et al., 1995). For PFGE, we used a CHEF Mapper™ with a cooling Module (Mini Chiller Model 1000) (Bio-Rad Hercules, CA 94547) containing permanently circulating 1× TAE buffer [4.84 g Tris-base (Sigma Chemical Co., St Louis, MO), 1.14 ml glacial acetic acid and 2 ml 0.5 M EDTA in 1 l deionized water]. We included molecular weight markers of chromosomal DNA of Hansenula wingei and Schizosaccharomyces pombe (Bio-Rad) in the PFGE agarose gel. CHEF electrophoresis was continued for a total migration time of 74 h at 2 V/cm with an angle of reorientation of ± 53° (106°) and a pulse time of 35 min. The temperature was maintained at 14°C by a minichiller (Bio-Rad).
Flow cytometry analysis
For each point, 106 cells were plated in DMEM and incubated for 48 h at 37°C. Cells were then incubated with replication inhibitors under the indicated conditions, in DMEM + 10% FCS at 37°C. Cells were then trypsinized, collected by centrifugation (5 min at 2000 g), re-suspended in 500 µl of PBS and fixed by adding 1.5 ml of cold ethanol. The DNA content was estimated by propidium iodide fluorescence and DNA flow cytometry (Becton FACScalibur).
Acknowledgments
Acknowledgements
We thank D.Rouillard (Institut Curie, Paris) for FACS expertise. We are grateful to Drs D.Marsh, F.Fabre and C.White for critical reading of the manuscript. Thanks are due to Drs M.Jasin, P.Jeggo and M.Liskay for providing us with CHO-DRA10, XR-1, xrs 6 and pJS3-10 cell lines, respectively. Y.S. was supported by an INSTN/EDF, then a FRM fellowship. S.L. and F.D. are supported by an INSTN fellowship. This work was supported by Electricité de France, ARC (9822 and 9238).
References
- Ausubel F., Brent,R., Kingston,R., Moore,D., Seidman,J., Smith,J. and Struhl,K. (1999) Current Protocols in Molecular Biology. Vols 1–4. John Wiley & Sons, Inc., Boston, MA.
- Bierne H., Ehrlich,S.D. and Michel,B. (1997) Deletions at stalled replication forks occur by two different pathways. EMBO J., 16, 3332–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligo M.A., Piras,A. and Rainaldi,G. (1988) Time course of sister chromatid exchanges and gene amplification induced by 1-β-d-arabinofuranosylcytosine in V79-AP4 Chinese hamster cells. Chromosoma, 96, 306–310. [DOI] [PubMed] [Google Scholar]
- Chen F., Nastasi,A., Shen,Z., Brenneman,M., Crissman,H. and Chen,D.J. (1997) Cell cycle-dependent protein expression of mammalian homologs of yeast DNA double-strand break repair genes Rad51 and Rad52. Mutat. Res., 384, 205–211. [DOI] [PubMed] [Google Scholar]
- Cox M.M., Goodman,M.F., Kreuzer,K.N., Sherratt,D.J., Sandler,S.J. and Marians,K.J. (2000) The importance of repairing stalled replication forks. Nature, 404, 37–41. [DOI] [PubMed] [Google Scholar]
- Dhermain F., Dardalhon,M., Queinnec,E. and Averbeck,D. (1995) Induction of double-strand breaks in Chinese hamster ovary cells at two different dose rates of γ-irradiation. Mutat. Res., 336, 161–167. [DOI] [PubMed] [Google Scholar]
- Dillehay L.E., Jacobson-Kram,D. and Williams,J.R. (1989) DNA topoisomerases and models of sister-chromatid exchange. Mutat. Res., 215, 15–23. [DOI] [PubMed] [Google Scholar]
- Dronkert M.L., Beverloo,H.B., Johnson,R.D., Hoeijmakers,J.H., Jasin,M. and Kanaar,R. (2000) Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol. Cell. Biol., 20, 3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers J., van Steeg,H., de Wit,J., Swagemakers,S.M., Vermeij,M., Hoeijmakers,J.H. and Kanaar,R. (2000) Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J., 19, 1703–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre F., Boulet,A. and Roman,H. (1984) Gene conversion at different points in the mitotic cycle of Saccharomyces cerevisiae. Mol. Gen. Genet., 195, 139–143. [DOI] [PubMed] [Google Scholar]
- Fram R.J. and Kufe,D.W. (1982) DNA strand breaks caused by inhibitors of DNA synthesis: 1-β-d-arabinofuranosylcytosine and aphidicolin. Cancer Res., 42, 4050–4053. [PubMed] [Google Scholar]
- Gunderson K. and Chu,G. (1991) Pulsed-field electrophoresis of megabase-sized DNA. Mol. Cell. Biol., 11, 3348–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaf T., Golub,E.I., Reddy,G., Radding,C.M. and Ward,D.C. (1995) Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl Acad. Sci. USA, 92, 2298–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber J.E. (1999) DNA repair. Gatekeepers of recombination. Nature, 398, 665–667. [DOI] [PubMed] [Google Scholar]
- Hyrien O. (2000) Mechanisms and consequences of replication fork arrest. Biochimie, 82, 5–17. [DOI] [PubMed] [Google Scholar]
- Ishii Y. and Bender,M.A. (1980) Effects of inhibitors of DNA synthesis on spontaneous and ultraviolet light-induced sister-chromatid exchanges in Chinese hamster cells. Mutat. Res., 79, 19–32. [DOI] [PubMed] [Google Scholar]
- Kadyk L.C. and Hartwell,L.H. (1992) Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics, 132, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein H.L. (1995) Genetic control of intrachromosomal recombination. BioEssays, 17, 147–159. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. (1995) Collapse and repair of replication forks in Escherichia coli. Mol. Microbiol., 16, 373–384. [DOI] [PubMed] [Google Scholar]
- Lambert S. and Lopez,B.S. (2000) Characterization of mammalian RAD51 double strand break repair using non lethal dominant negative forms. EMBO J., 19, 3090–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S., Saintigny,Y., Delacote,F., Amiot,F., Chaput,B., Lecomte,M., Huck,S., Bertrand,P. and Lopez,B.S. (1999) Analysis of intrachromosomal homologous recombination in mammalian cell, using tandem repeat sequences. Mutat. Res., 433, 159–168. [DOI] [PubMed] [Google Scholar]
- Levenson V. and Hamlin,J.L. (1993) A general protocol for evaluating the specific effects of DNA replication inhibitors. Nucleic Acids Res., 21, 3997–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F., Han,M., Romanienko,P.J. and Jasin,M. (1998) Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl Acad. Sci. USA, 95, 5172–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liskay R.M., Stachelek,J.L. and Letsou,A. (1984) Homologous recombination between repeated chromosomal sequences in mouse cells. Cold Spring Harb. Symp. Quant. Biol., 49, 183–189. [DOI] [PubMed] [Google Scholar]
- Morita T., Yoshimura,Y., Yamamoto,A., Murata,K., Mori,M., Yamamoto,H. and Matsushiro,A. (1993) A mouse homolog of the Escherichia coli recA and Saccharomyces cerevisiae RAD51 genes. Proc. Natl Acad. Sci. USA, 90, 6577–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive P.L., Banath,J.P. and Durand,R.E. (1990) Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the ‘comet’ assay. Radiat. Res., 122, 86–94. [PubMed] [Google Scholar]
- Rattray A.J. and Symington,L.S. (1995) Multiple pathways for homologous recombination in Saccharomyces cerevisiae. Genetics, 139, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein R., Michel,B. and Gangloff,S. (2000) Replication fork pausing and recombination or ‘gimme a break’. Genes Dev., 14, 1–10. [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Scully R., Puget,N. and Vlasakova,K. (2000) DNA polymerase stalling, sister chromatid recombination and the BRCA genes. Oncogene, 19, 6176–6183. [DOI] [PubMed] [Google Scholar]
- Seigneur M., Bidnenko,V., Ehrlich,S.D. and Michel,B. (1998) RuvAB acts at arrested replication forks. Cell, 95, 419–430. [DOI] [PubMed] [Google Scholar]
- Shinohara A., Ogawa,H., Matsuda,Y., Ushio,N., Ikeo,K. and Ogawa,T. (1993) Cloning of human, mouse and fission yeast recombination genes homologous to RAD51 and recA [published erratum appears in Nature Genet., 1993, 5, 312]. Nature Genet., 4, 239–243. [DOI] [PubMed] [Google Scholar]
- Siede W., Friedl,A.A., Dianova,I., Eckardt-Schupp,F. and Friedberg,E.C. (1996) The Saccharomyces cerevisiae Ku autoantigen homologue affects radiosensitivity only in the absence of homologous recombination. Genetics, 142, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.P., McCoy,M.T., Tice,R.R. and Schneider,E.L. (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res., 175, 184–191. [DOI] [PubMed] [Google Scholar]
- Sonoda E., Sasaki,M.S., Buerstedde,J.M., Bezzubova,O., Shinohara,A., Ogawa,H., Takata,M., Yamaguchi-Iwai,Y. and Takeda,S. (1998) Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J., 17, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E., Sasaki,M.S., Morrison,C., Yamaguchi-Iwai,Y., Takata,M. and Takeda,S. (1999) Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol. Cell. Biol., 19, 5166–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M., Sasaki,M.S., Sonoda,E., Morrison,C., Hashimoto,M., Utsumi,H., Yamaguchi-Iwai,Y., Shinohara,A. and Takeda,S. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J., 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki T., Fujii,Y., Sakumi,K., Tominaga,Y., Nakao,K., Sekiguchi,M., Matsushiro,A., Yoshimura,Y. and Morita,T. (1996) Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl Acad. Sci. USA, 93, 6236–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck E., Stasiak,A.Z., Stasiak,A. and West,S.C. (1999) Binding of double-strand breaks in DNA by human Rad52 protein. Nature, 398, 728–731. [DOI] [PubMed] [Google Scholar]
- Vispé S., Cazaux,C., Lesca,C. and Defais,M. (1998) Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res., 26, 2859–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A. et al. (1996) Cell cycle-dependent expression of the mouse Rad51 gene in proliferating cells. Mol. Gen. Genet., 251, 1–12. [DOI] [PubMed] [Google Scholar]
- Zou H. and Rothstein,R. (1997) Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell, 90, 87–96. [DOI] [PubMed] [Google Scholar]