




Abstract
T cell stimulation via glucocorticoid-induced tumor necrosis factor receptor family–related protein (GITR) can evoke effective tumor immunity. A single administration of agonistic anti-GITR monoclonal antibody (mAb) to tumor-bearing mice intravenously or directly into tumors provoked potent tumor-specific immunity and eradicated established tumors without eliciting overt autoimmune disease. A large number of CD4+ and CD8+ T cells, including interferon (IFN)-γ–secreting cells, infiltrated regressing tumors. Tumor-specific IFN-γ–secreting CD4+ and CD8+ T cells also increased in the spleen. The treatment led to tumor rejection in IFN-γ–intact mice but not IFN-γ–deficient mice. Furthermore, coadministration of anti-GITR and anti–CTLA-4 mAbs had a synergistic effect, leading to eradication of more advanced tumors. In contrast, coadministration of anti-CD25 and anti-GITR mAbs was less effective than anti-GITR treatment alone, because anti-CD25 depleted both CD25+-activated effector T cells and CD25+CD4+ naturally occurring regulatory T (T reg) cells. Importantly, CD4+ T cells expressing the T reg–specific transcription factor Foxp3 predominantly infiltrated growing tumors in control mice, indicating that tumor-infiltrating natural Foxp3+CD25+CD4+ T reg cells may hamper the development of effective tumor immunity. Taken together, T cell stimulation through GITR attenuates T reg–mediated suppression or enhances tumor-killing by CD4+ and CD8+ effector T cells, including those secreting IFN-γ, or both. Agonistic anti-GITR mAb is therefore instrumental in treating advanced cancers.
There is substantial evidence that cancer patients harbor tumor-reactive T cells, although their reactivity or number is usually insufficient to eradicate tumors (1). How such tumor-reactive T cells can be sufficiently activated and expanded to cure established tumors is a key issue for devising effective immunotherapy for cancer (2). One way of achieving this is to breach the mechanisms of peripheral self-tolerance that may hamper the activation of T cells reactive with tumor-associated antigens, many of which are normal self-antigens (1). There is accumulating evidence that naturally occurring CD25+CD4+ regulatory T (T reg) cells not only engage in the maintenance of immunologic self-tolerance in the periphery but also impede immunosurveillance against autologous tumor cells (3). For example, depletion of CD25+CD4+ T cells by administration of anti-CD25 mAb before tumor challenge provokes effective immune responses to syngeneic tumors in otherwise nonresponding animals (4–6). In humans, tumor-reactive T cells can be efficiently expanded in vitro when CD25+CD4+ T cells are depleted from PBMCs before stimulation with tumor-derived peptide (7). A key issue in tumor immunology is then to determine how effective immune responses against advanced tumors can be provoked by attenuating T reg–mediated suppression and, concomitantly, stimulating tumor-reactive T cells present in cancer-bearing hosts.
CD25+CD4+ natural T reg cells constitutively express the transcription factor Foxp3, cytotoxic T lymphocyte-associated protein 4 (CTLA-4), and glucocorticoid-induced TNF receptor family-related protein (GITR) (TNFRSF18) (8–15). They express GITR at higher levels than other T cells, although both T reg and non–T reg cells up-regulate its expression upon activation (11, 12). In vitro studies have shown that cross-linking of GITR, not its blockade, by a specific mAb, together with TCR stimulation, abrogates CD25+CD4+ T cell–mediated suppression, triggers proliferation of T reg cells in the presence of interleukin 2, and exhibits costimulatory activity for TCR-stimulated T cell activation (11, 12, 16–19). Administration of the mAb to neonatal mice can indeed break self-tolerance and elicit autoimmune disease (11). This GITR-mediated attenuation of suppression and costimulation of effector T cells synergistically enhanced in vivo antigen-specific immune responses such as antiviral immunity, allograft rejection, and graft-versus-host reaction (20–22). We examined the immunostimulatory activity of agonistic anti-GITR mAb to provoke effective tumor immunity in mice with advanced tumors. We also assessed local and systemic effects of mAb on tumor-targeting effector T cells and Foxp3-expressing T reg cells; its possible synergy with other mAbs, such as anti–CTLA-4, to further enhance tumor immunity; and possible autoimmune-inducing effects of these mAbs in treated animals.
RESULTS AND DISCUSSION
Eradication of established tumors by agonistic anti-GITR mAb but not by cell-depleting anti-CD25 mAb
DTA-1 is a rat mAb of IgG2b isotype and is incapable of depleting GITR-expressing cells in vivo (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20050940/DC1) (11). Meth A, a BALB/c-derived fibrosarcoma cell line, does not express GITR—in contrast with hematopoietic tumor lines, many of which express GITR (Fig. S1 B)—and DTA-1 treatment did not affect the growth of Meth A in athymic nude mice (Fig. S1 C). To determine whether DTA-1 can evoke effective tumor immunity, we injected 500 μg of DTA-1 intravenously on various days after intradermal inoculation of Meth A to normal BALB/c mice (Fig. 1 A). One-shot DTA-1 injection between days 0 and 12 after tumor inoculation led to tumor regression. The injection on day 8, when tumors were already palpable, was most effective, and nearly 90% of such mice rejected tumors. As for dose response, 100 or 20 μg DTA-1 injection on day 8 led to tumor eradication in 70% (4/7) and 14% (1/7) of mice, respectively. Multiple injections were more effective than a single injection (unpublished data). The results contrasted with the antitumor effect of PC61 anti-CD25 mAb, which is of the rat IgG1 isotype and cell-depleting in vivo (4, 5) (Fig. S1 A); that is, PC61 injection 4 d before tumor inoculation was effective in provoking tumor regression, whereas injection on day 0 or thereafter was ineffective (Fig. 1 A).
Figure 1.

Tumor immunity induced by anti-GITR mAb treatment. (A) BALB/c mice 8–10 wk of age were inoculated intradermally with 2 × 105 Meth A on their back on day 0. Five hundred micrograms of DTA-1, PC61, or control rat IgG was injected intravenously on the indicated days, and the tumor size of each mouse was monitored every other day. (B) BALB/c mice were intradermally inoculated with 105 Colon 26 cells on day 0 and injected intravenously with 500 μg DTA-1 on day 8. Control mice were injected with rat IgG on day 8. (C) BALB/c mice that had completely rejected Meth A following DTA-1 treatment on day 8 were intradermally inoculated on day 70 with a 10-fold larger dose of Meth A (2×106 cells) and Colon 26 (1×105 cells) on either side of the back, and monitored for tumor growth. *P < 0.001 via one-sided Fisher exact probability test. Results are representative of three independent experiments.
DTA-1 injection on day 8 also led to regression of Colon 26, a GITR-nonexpressing BALB/c-derived rectal carcinoma cell line, at a significant rate (Fig. 1 B). The treatment was similarly effective on other tumor lines, including GITR-expressing hematopoietic tumor lines (unpublished data). It was much less effective, however, on other aggressive tumor lines such as B16 melanoma in B6 mice (unpublished data), although DTA-1 treatment of B16 could induce effective concomitant immunity (23).
Once DTA-1–treated BALB/c mice rejected Meth A, they rapidly rejected a subsequent challenge with a 10-fold larger dose of Meth A but failed to reject Colon 26 inoculated at the same time at another site even at the cell dose leading to tumor rejection after anti-GITR treatment (Fig. 1 C), indicating that these mice had become specifically immune to Meth A.
Thus, DTA-1 treatment of tumor-bearing mice can evoke tumor-specific immunity that is sufficiently potent to eradicate established tumors.
Intratumor infiltration of CD4+ and CD8+ T cells after anti-GITR treatment and predominant infiltration of Foxp3+CD4+ T cells in control tumors
Immunohistochemical characterization of T cells in Meth A tumors in BALB/c mice treated with DTA-1 on day 8 revealed a large number of infiltrating CD4+ and CD8+ T cells (Fig. 2, A and B). In contrast, a much smaller number of CD4+ or CD8+ T cells infiltrated growing tumors in control mice; the majority of these infiltrating CD4+ cells expressed Foxp3, especially in the early days after tumor inoculation.
Figure 2.
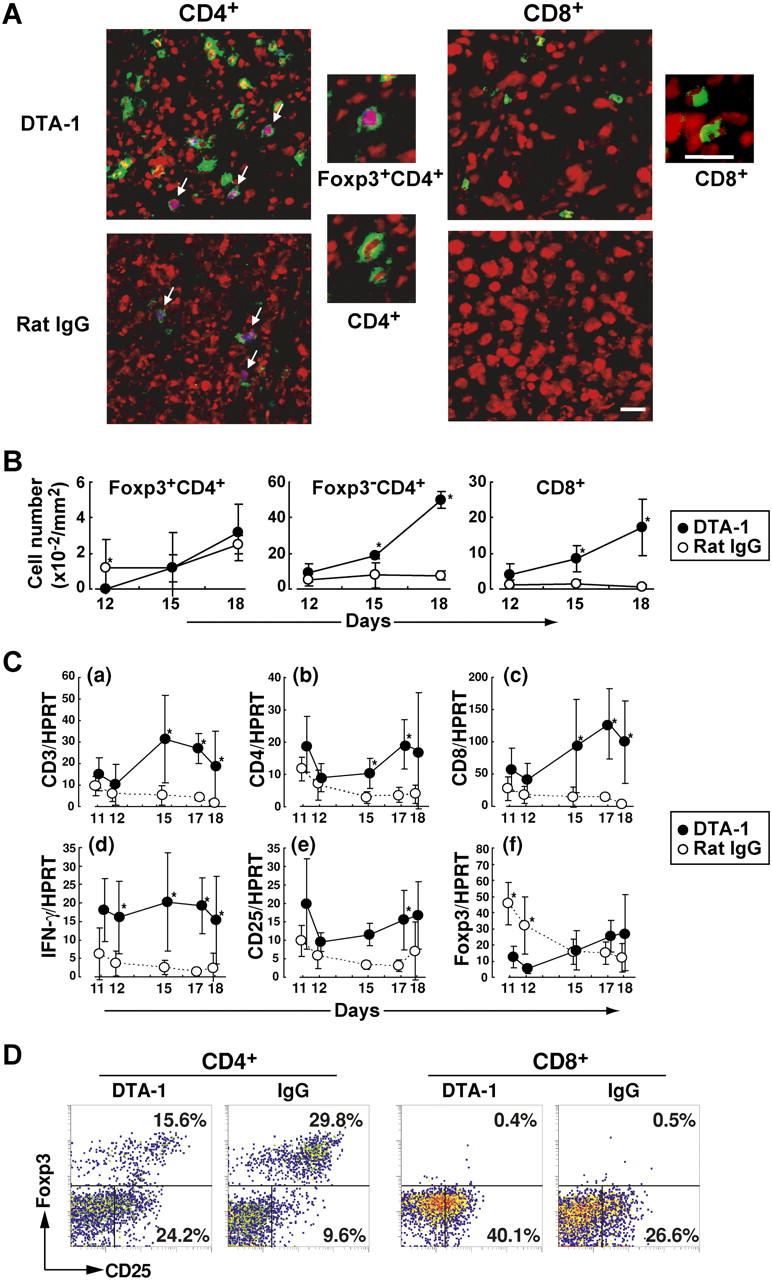
Tumor infiltration of CD4+ and CD8+ T cells, IFN-γ–secreting cells, and Foxp3-expressing T reg cells. (A) Mice were treated with either DTA-1 or rat IgG on day 8 after tumor inoculation and their tumor tissues were taken on day 18 and stained with anti-CD4 or anti-CD8 (green), anti-Foxp3 and Alexa633-anti–rabbit IgG (blue), and POPO-3 iodide for nuclear staining (red), hence purple nuclear staining of Foxp3-expressing cells (arrows). Scale bars: 20 μm. (B) The number of each T cell population per visual field (200 × 200 μm) was counted under a microscope, and the average numbers per 1 mm2 in five sections of each tumor (n = 3 on each time point) obtained on the indicated days are shown. (C) RNA was extracted from each tumor tissue (n = 5–8) obtained on indicated days after tumor inoculation. With each specimen, expression levels of mRNA for CD3, CD4, CD8, IFN-γ, CD25, Foxp3, and HPRT were assessed by quantitative real-time PCR, and standardized with HPRT expression levels (a–f). Ordinates are arbitrary units. *P < 0.05 via two-sided t test. (D) Tumor-infiltrating cells in DTA-1– or IgG-treated mice on day 18 after tumor inoculation, as in (A), were stained with anti-Foxp3 and anti-CD25, and gated for CD4+ or CD8+ cells. Results are representative of three independent experiments.
Quantitative assessment of messenger RNA (mRNA) encoding various genes by real-time polymerase chain reaction (PCR) revealed that messages for CD3+, CD4+, CD8+, CD25+, and IFN-γ+ cells per tumor volume were significantly higher in DTA-1–treated tumors than controls (Fig. 2 C). In contrast, Foxp3 message was higher in control tumors in the early phase of tumor growth, showing a good correlation between the number (Fig. 2 B) and mRNA message (Fig. 2, C–F). The ratios of the messages for Foxp3 to CD3 were also much higher in growing tumors of control mice than in DTA-treated tumors, even in the later phase (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20050940/DC1). Intracellular staining of the Foxp3 protein at a single cell level also revealed that, on day 18 after tumor inoculation, Foxp3+ cells were lower in ratio among tumor-infiltrating CD4+ T cells in DTA-1–treated mice compared with control mice, whereas Foxp3−CD25+ cells were much higher in the former. CD25+CD8+ cells also infiltrated to a much higher ratio in the former.
Taken together, these results indicate that DTA-1 treatment induces tumor infiltration of CD4+ and CD8+ T cells, including activated CD25+ T cells and IFN-γ–forming cells, whereas Foxp3+CD25+CD4+ T reg cells predominantly infiltrate nontreated tumors without much accompanying infiltration of activated T cells.
Requirement of IFN-γ for tumor eradication after anti-GITR treatment
To determine whether DTA-1 treatment of tumor-bearing mice induces IFN-γ–secreting cells in the spleen as well, spleen cells from BALB/c mice either Meth A–inoculated or noninoculated and subsequently DTA-1–treated or nontreated were cultured in vitro with or without Meth A for 18 h, and the number of IFN-γ–secreting cells was assessed (Fig. 3 A). Spleens from Meth A–inoculated DTA-1–treated mice developed significantly larger numbers of IFN-γ–secreting cells than those from Meth A–inoculated but DTA-1–nontreated mice whether or not the spleen cells were stimulated in vitro with Meth A. These IFN-γ–secreting cells appeared to be tumor-specific, because in vitro stimulation with other tumor cells failed to enhance IFN-γ secretion by Meth-A/DTA-1–treated mouse spleen cells (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20050940/DC1); they were CD4+ or CD8+, but not CD4− or CD8− (Fig. 3 B). Importantly, DTA-1 treatment alone failed to induce them, indicating requirement of antigen sensitization for their development. Furthermore, IFN-γ–intact BALB/c mice treated with DTA-1 on day 8 rejected tumors, whereas similarly treated IFN-γ–deficient BALB/c mice did not (Fig. 3 C). These results collectively indicate that DTA-1 treatment increases the number of tumor-specific CD4+ and CD8+ T cells secreting IFN-γ, which appears to be indispensable for tumor rejection in this system (24, 25). Given a variety of antitumor activities of IFN-γ through promoting innate and adaptive immune responses (26), further study is required to determine how natural T reg cells physiologically control the development of IFN-γ–secreting tumor-specific effector T cells.
Figure 3.
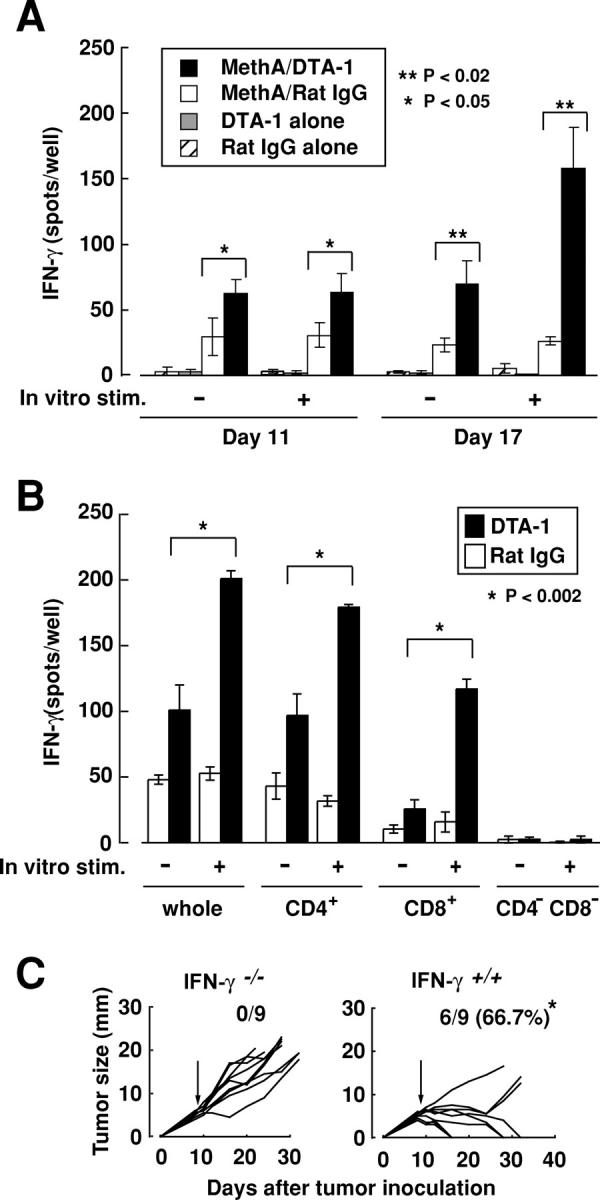
Requirement of IFN-γ–secreting effector T cells for tumor rejection in anti-GITR mAb–treated mice. (A) Whole splenocytes from four groups either Meth A–inoculated or not on day 0, or DTA-1–treated or not on day 8 were taken on day 11 and 17, and cultured with or without Meth A for 18 h. The numbers of IFN-γ–secreting cells were determined by ELISPOT assay. Data are represented as the mean ± SD of 3 mice each. Statistical analyses were performed using a two-sided t test. (B) CD4+, CD8+, and CD4−CD8− splenic T cells from Meth A–inoculated and DTA-1–treated mice (n = 4) were prepared on day 17, cultured with or without Meth A, and assessed for the number of IFN-γ–secreting cells. Data are shown as the mean ± SD of triplicate wells. (C) 6–8-wk-old normal naive BALB/c mice or IFN-γ–deficient mice were inoculated with 2 × 105 Meth A intradermally on their back on day 0 and injected intravenously with 500 μg of DTA-1 on day 8. *P < 0.04 via one-sided Fisher exact probability test.
Eradication of advanced tumors by a combination of anti-GITR and anti–CTLA-4 treatment or intratumor injection of anti-GITR
As reported previously and shown in Fig. 1, administration of anti-CD25 and anti–CTLA-4 mAbs can enhance tumor immunity (4–6). Coadministration of DTA-1 and PC61 on day 8, however, reduced the tumor-eradicating efficacy of DTA-1, indicating that PC61 depletes CD25+ activated effector T cells as well as natural CD25+CD4+ T reg cells (Fig. 4 A). This correlates with the infiltration of both populations into tumors (Fig. 2) and successful tumor rejection only by PC61 treatment before tumor inoculation, but not after (Fig. 1 A). In contrast, coadministration of DTA-1 and 4F10—a nondepleting hamster anti–CTLA-4 mAb of the IgG isotype (8, 27)—on day 12 when either mAb alone was only marginally effective for tumor eradication successfully induced tumor regression in a significant number of mice (Fig. 4 B). In addition, an intratumor injection of a small dose (50 μg) of DTA-1 on day 12 was highly effective in tumor rejection (Fig. 4 C). Thus anti–CTLA-4 is synergistic with anti-GITR in provoking tumor immunity, leading to eradication of more advanced tumors. Furthermore, local as well as systemic administration of DTA-1 is effective in evoking tumor immunity.
Figure 4.

Synergistic antitumor effect of anti-GITR and anti-CTLA-4, but not anti-GITR and anti-CD25. (A) BALB/c mice inoculated intradermally with 2 × 105 Meth A were injected intravenously on day 8 with 500 μg DTA-1, 500 μg PC61, a mixture of the two, or 500 μg rat IgG. (B) Tumor-inoculated BALB/c mice were similarly injected on day 12 with 500 μg DTA-1, 500 μg 4F10, or a mixture of the two. (C) Tumor inoculated BALB/c mice received an intratumor injection of 50 μg of DTA-1 or rat IgG in 10 μL on day 12 after intradermal inoculation of 2 × 105 Meth A. *P < 0.001 via one-sided Fisher exact probability test. Results are representative of three independent experiments.
Failure to elicit overt autoimmune disease by anti-GITR mAb treatment
As previously shown, neonatal injection of 1 mg 4F10 or DTA-1 on day 10 and 20 after birth can induce histologically and serologically overt autoimmune disease (e.g., autoimmune gastritis accompanying antiparietal cell autoantibody in BALB/c mice) (10, 11). In the present experiments, administration of an equivalent amount of DTA-1, or a mixture of DTA-1 and 4F10, to adult BALB/c mice induced potent tumor immunity but failed to produce histologically evident autoimmune gastritis or other autoimmune diseases, although the treated mice, especially those surviving >3 mo, developed low to moderate titers of antiparietal cell autoantibody (Fig. 5). Intratumor inoculation proved weaker in the induction of autoantibodies, with hardly any being detectable.
Figure 5.

No overt autoimmune disease after mAb treatment. BALB/c mice treated as shown in Figs. 1 and 4, and those that survived indicated days were serologically and histologically assessed for the development of autoimmune gastritis and circulating antiparietal cell autoantibody. In all cases, there was a histologically intact gastric mucosa.
The present findings have the following implications. First, agonistic GITR antibody can provoke potent tumor-specific immunity in tumor-bearing mice. Although the precise molecular mechanism of this potentiation remains to be determined, it is likely that, in the presence of antigenic stimulation, the antibody attenuates T reg–mediated suppression, enhances the activity of effector T cells to the degree sufficient to overcome the suppression, renders them resistant to the suppression, or exerts a combination of these effects (11, 12, 16–19). Anti–CTLA-4, especially blocking antibody, appears to have a similar effect; that is, attenuation of T reg–mediated suppression by blocking the activation of T reg cells, enhancement of effector activity by blocking negative signal to effector T cells, or both (8–10, 27, 28). These mAbs can therefore act synergistically in enhancing immune responses such as tumor immunity. Second, Foxp3-expressing CD25+CD4+ natural T reg cells may preferentially infiltrate into tumors and impede antitumor immune responses in the tumors, presumably in the regional lymph nodes and spleen (29). Given the high self-reactivity of T reg cells, they are recruited because they recognize not only tumor-associated antigens but also normal self-antigens released from malignant or normal cells damaged in the tumor (3). Monitoring such tumor-infiltrating Foxp3+ T reg cells is of help in assessing the status of host tumor immunity. Third, anti-GITR and, for that matter, anti-CTLA-4 treatment, both of which are potentially capable of triggering autoimmunity, can evoke effective tumor immunity without overt autoimmune disease. This differential effect mainly depends on the dose of agonistic anti-GITR antibody, the route (systemic or local) of administration, the duration of the treatment, the age of the treated host, and its genetic susceptibility to autoimmune disease (3, 30). Practically, a combination of tumor vaccination with local manipulation of T reg cells, for example, via intratumor inoculation of agonistic anti-GITR mAb or immunization with tumor-derived antigens along with the mAb, may make the current cancer immunotherapy more efficacious without deleterious autoimmunity. The approach used here could also potentially be employed for effective vaccination against microbes.
MATERIALS AND METHODS
Mice.
Female BALB/c mice 8–10 wk of age were purchased from Japan SLC. BALB/c IFN-γ−/− mice were purchased from The Jackson Laboratory. They were maintained in our animal facility and treated in accordance with the guidelines for animal care approved by the Institute for Frontier Medical Sciences, Kyoto University. Tumor-inoculated mice were killed when average tumor diameters reached 20 mm or there was no tendency of tumor regression 30 d after tumor inoculation.
Tumor cells.
Meth A was a gift from E. Nakayama (Okayama University, Okayama, Japan). Colon 26 was obtained from Cell Resource Center for Biomedical Research, Tohoku University.
Antibodies.
FITC–anti-CD25 (7D4) and anti–CD4-PE were purchased from PharMingen. Anti-CD25 (PC61) (rat IgG1) secreting hybridoma was purchased from American Type Culture Collection. The hybridoma cells secreting anti-CTLA-4 mAb (UC10-4F10-11) were a gift from J. Bluestone (University of California, San Francisco, CA). R-PE-Cy5-conjugated streptavidin (Dako/Japan) was used as the secondary reagent for biotinylated antibodies. PC61, DTA-1, and 4F10 were purified from ascites of SCID mice by 40% ammonium sulfate precipitation twice. DTA-1 was subsequently purified by protein G column (GE Healthcare). Purified rat IgG and hamster IgG were purchased from Sigma and Cappel, respectively. Rabbit anti-mouse Foxp3 was prepared by immunizing a rabbit with Foxp3-transfected WEHI cells (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20050940/DC1). PE-labeled anti–mouse Foxp3 mAb was purchased from e-BioScience, and used according to the manufacturer's instructions.
Serologic analysis and histology.
Detailed descriptions of the ELISA for detecting autoantibodies against the gastric parietal cell antigens was previously described (12). IFN-γ ELISPOT assay was performed using the Cytokine ELISPOT set (BD Biosciences) according to the manufacturer's instruction. 5 × 105 freshly isolated whole splenocytes, CD4−CD8− splenocytes, CD8+ and CD8− T cells prepared by MACS (Miltenyi Biotec) were incubated with or without 2 × 104 mitomycin C-treated Meth A on the coated membranes for 18 h at 37°C under 5% CO2. The spots were automatically counted using KS ELISPOT compact (Carl Zeiss).
Immunohistochemistry.
Cryostat-frozen sections were dried and fixed with cold acetone for 15 min at 4°C. Sections were permeabilized with phosphate-buffered saline containing 0.2% Tween 20, incubated with phosphate-buffered saline containing 1% bovine serum albumin for 30 min at room temperature, then with rabbit anti–mouse Foxp3 Ab and rat anti–mouse CD4 mAb (H129.19; BD Biosciences) or rat anti–CD8 (53–6.7; BD Biosciences) for 1 h at room temperature, and subsequently with Alexa488-conjugated goat anti–rat IgG, Alexa633-conjugated goat anti–rabbit IgG and POPO-3 iodide (Invitrogen), and examined with an LSM 510 META microscope (Carl Zeiss).
Quantitative PCR.
Real-time quantitative PCR was performed as previously described (14). To exclude amplification from genomic DNA contamination, either the primers or the probes were designed to overlap splice junction. All probes but for HPRT were labeled with the fluorescent dyes 5′-FAM (6-carboxy-fluorescein) as reporter and 3′-TAMRA (6-carboxy-tetramethyl-rhodamine) as quencher. Probe for HPRT amplification was 5′-VIC labeled. The sequences of forward primers, Taq Man probes (28), and reverse primers, respectively, were as follows: Foxp3, 5′-CCCAGGAAAGACAGCAACCTT-3′, 5′-ATCCTACCCACTGCTGGCAAATGGAGTC-3′, 5′-TTCTCACAAGGCCACTTG-3′; CD8β, 5′-GAAGCAATGCCCGTTCCC-3′, 5′-ACCCAGAGACCCAGAAGGGCCTGAC-3′, 5′-TGAGGGTGGTAAGGCTACA-3′; CD4, 5′-CAGCATGGCAAAGGTGTATTAATTAG-3′, 5′-AGGTTCGCCTTCGCAGTTTGATCGT-3′, 5′-CCCATGCCCCTTTTTTGG-3′; CD3ɛ, 5′-CCACCTGTTCCCAACCCAG-3′, 5′-TGAGCCCATCCGCAAAGGCC-3′, 5′-TCAGGCCAGAATACAGGTCCC-3′; CD25, 5′-AGACTTCCTGCCCCATAACCA-3′, 5′-CACAGACTTCCCACAACCCACAGAAACAAC-3′, 5′-TGAGCACAAATGTCTCCGTAC-3′; and HPRT, 5′-TGAAGAGCTACTGTAATGATCAGTCAAC-3′, 5′-TGCTTTCCCTGGTTAAGCAGTACAGCCC-3′, 5′-AGCAAGCTTGCAACCTTAACCA-3′.
Statistical analysis.
All statistical analyses were performed using the Student's paired t test or Fisher exact probability test.
Online supplemental material.
Fig. S1 shows expression of GITR on various tumor lines, effects of DTA-1 and PC61 on CD25+CD4+ T cells in normal mice, and effects of DTA-1 on the growth of Meth A tumors inoculated in athymic nude mice. Fig. S2 shows the ratios of Foxp3/CD3 mRNA message in DTA-1–treated or control tumors on various days after treatment. Fig. S3 shows tumor specificity of IFN-γ–secreting spleen cells from tumor-inoculated and DTA-1–treated mice. Fig. S4 shows the specificity of rabbit anti-Foxp3 polyclonal Ab used in the present experiments. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050940/DC1.
Acknowledgments
We thank Z. Fehervari for critically reading the manuscript, and T. Matsushita for histology.
This work was supported by grants-in-aid from the Ministry of Education, Sports, and Culture of Japan.
The authors have no conflicting financial interests.
References
- 1.Van Der Bruggen, P., Y. Zhang, P. Chaux, V. Stroobant, C. Panichelli, E.S. Schultz, J. Chapiro, B.J. Van Den Eynde, F. Brasseur, and T. Boon. 2002. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol. Rev. 188:51–64. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg, S.A., J.C. Yang, and N.P. Restifo. 2004. Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 10:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakaguchi, S. 2004. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22:531–562. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu, J., S. Yamazaki, and S. Sakaguchi. 1999. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J. Immunol. 163:5211–5218. [PubMed] [Google Scholar]
- 5.Onizuka, S., I. Tawara, J. Shimizu, S. Sakaguchi, T. Fujita, and E. Nakayama. 1999. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 59:3128–3133. [PubMed] [Google Scholar]
- 6.Sutmuller, R.P., L.M. van Duivenvoorde, A. van Elsas, T.N. Schumacher, M.E. Wildenberg, J.P. Allison, R.E. Toes, R. Offringa, and C.J. Melief. 2001. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 194:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danke, N.A., D.M. Koelle, C. Yee, S. Beheray, and W.W. Kwok. 2004. Autoreactive T cells in healthy individuals. J. Immunol. 172:5967–5972. [DOI] [PubMed] [Google Scholar]
- 8.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]
- 9.Read, S., V. Malmstrom, and F. Powrie. 2000. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi, T., T. Tagami, S. Yamazaki, T. Uede, J. Shimizu, N. Sakaguchi, T. W. Mak, and S. Sakaguchi. 2000. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimizu, J., S. Yamazaki, T. Takahashi, Y. Ishida, and S. Sakaguchi. 2002. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3:135–142. [DOI] [PubMed] [Google Scholar]
- 12.McHugh, R.S., M.J. Whitters, C.A. Piccirillo, D.A. Young, E.M. Shevach, M. Collins, and M.C. Byrne. 2002. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 16:311–323. [DOI] [PubMed] [Google Scholar]
- 13.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 14.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 15.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 16.Kanamaru, F., P. Youngnak, M. Hashiguchi, T. Nishioka, T. Takahashi, S. Sakaguchi, I. Ishikawa, and M. Azuma. 2004. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J. Immunol. 172:7306–7314. [DOI] [PubMed] [Google Scholar]
- 17.Stephens, G.L., R.S. McHugh, M.J. Whitters, D.A. Young, D. Luxenberg, B.M. Carreno, M. Collins, and E.M. Shevach. 2004. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J. Immunol. 173:5008–5020. [DOI] [PubMed] [Google Scholar]
- 18.Ji, H.B., G. Liao, W.A. Faubion, A.C. Abadia-Molina, C. Cozzo, F.S. Laroux, A. Caton, and C. Terhorst. 2004. Cutting edge: the natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J. Immunol. 172:5823–5827. [DOI] [PubMed] [Google Scholar]
- 19.Ronchetti, S., O. Zollo, S. Bruscoli, M. Agostini, R. Bianchini, G. Nocentini, E. Ayroldi, and C. Riccardi. 2004. Frontline: GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur. J. Immunol. 34:613–622. [DOI] [PubMed] [Google Scholar]
- 20.Wood, K.J., H. Ushigome, M. Karim, A. Bushell, S. Hori, and S. Sakaguchi. 2003. Regulatory cells in transplantation. Novartis Found. Symp. 252:177–188. [PubMed] [Google Scholar]
- 21.Dittmer, U., H. He, R.J. Messer, S. Schimmer, A.R. Olbrich, C. Ohlen, P.D. Greenberg, I.M. Stromnes, M. Iwashiro, S. Sakaguchi, et al. 2004. Functional impairment of CD8+ T cells by regulatory T cells during persistent retroviral infection. Immunity. 20:293–303 [erratum: Immunity. 20:653]. [DOI] [PubMed] [Google Scholar]
- 22.Muriglan, S.J., T. Ramirez-Montagut, O. Alpdogan, T.W. Van Huystee, J.M. Eng, V.M. Hubbard, A.A. Kochman, K.H. Tjoe, C. Riccardi, P.P. Pandolfi, et al. 2004. GITR activation induces an opposite effect on alloreactive CD4+ and CD8+ T cells in graft-versus-host disease. J. Exp. Med. 200:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turk, M.J., J.A. Guevara-Patino, G.A. Rizzuto, M.E. Engelhorn, S. Sakaguchi, and A.N. Houghton. 2005. 2004. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J. Exp. Med. 200:771–782 [erratum: J. Exp. Med. 201:159]. [DOI] [PMC free article] [PubMed]
- 24.Dighe, A.S., E. Richards, L.J. Old, and R.D. Schreiber. 1994. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1:447–456. [DOI] [PubMed] [Google Scholar]
- 25.Casares, N., L. Arribillaga, P. Sarobe, J. Dotor, A. Lopez-Diaz de Cerio, I. Melero, J. Prieto, F. Borras-Cuesta, and J.J. Lasarte. 2003. CD4+/CD25+ regulatory cells inhibit activation of tumor-primed CD4+ T cells with IFN-γ-dependent antiangiogenic activity, as well as long-lasting tumor immunity elicited by peptide vaccination. J. Immunol. 171:5931–5939. [DOI] [PubMed] [Google Scholar]
- 26.Ikeda, H., L.J. Old, and R.D. Schreiber. 2002. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 13:95–109. [DOI] [PubMed] [Google Scholar]
- 27.Tang, Q., E.K. Boden, K.J. Henriksen, H. Bour-Jordan, M. Bi, and J.A. Bluestone. 2004. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34:2996–3005. [DOI] [PubMed] [Google Scholar]
- 28.Chambers, C.A., M.S. Kuhns, J.G. Egen, and J.P. Allison. 2001. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 19:565–594. [DOI] [PubMed] [Google Scholar]
- 29.Yu, P., Y. Lee, W. Liu, T. Krausz, A. Chong, H. Schreiber, and Y.X. Fu. 2005. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J. Exp. Med. 201:779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phan, G.Q., J.C. Yang, R.M. Sherry, P. Hwu, S.L. Topalian, D.J. Schwartzentruber, N.P. Restifo, L.R. Haworth, C.A. Seipp, L.J. Freezer, et al. 2003. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA. 100:8372–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]