




Abstract
The mapping of quantitative trait loci (QTL) is to identify molecular markers or genomic loci that influence the variation of complex traits. The problem is complicated by the facts that QTL data usually contain a large number of markers across the entire genome and most of them have little or no effect on the phenotype. In this article, we propose several Bayesian hierarchical models for mapping multiple QTL that simultaneously fit and estimate all possible genetic effects associated with all markers. The proposed models use prior distributions for the genetic effects that are scale mixtures of normal distributions with mean zero and variances distributed to give each effect a high probability of being near zero. We consider two types of priors for the variances, exponential and scaled inverse-χ2 distributions, which result in a Bayesian version of the popular least absolute shrinkage and selection operator (LASSO) model and the well-known Student's t model, respectively. Unlike most applications where fixed values are preset for hyperparameters in the priors, we treat all hyperparameters as unknowns and estimate them along with other parameters. Markov chain Monte Carlo (MCMC) algorithms are developed to simulate the parameters from the posteriors. The methods are illustrated using well-known barley data.
QUANTITATIVE traits are controlled by multiple quantitative trait loci (QTL). The genetic effects of QTL and the phenotypic value of a quantitative trait are usually described by a linear model. Since the locations of QTL are not known a priori, we often use markers to represent QTL. Some markers may be closely linked to one or more QTL, and thus they may show strong association with the trait. Most markers, however, may not be directly linked to QTL, and thus no association will be expected between these markers and the trait. Interval mapping or the equivalent single-marker analysis does not provide accurate estimates of QTL effects because the single-QTL model used by the method is seldom the correct model. The multiple-QTL model is the reasonable choice for mapping quantitative traits (Kao et al. 1999).
When all markers are included in the QTL analysis, the model may be oversaturated. Variable selection may be conducted to include and exclude markers in the model (Sillanpää and Arjas 1998; Yi et al. 2003, 2005; Yi 2004; Yi and Shriner 2008). An alternative approach is a shrinkage method that includes all variables in the model and uses informative prior distributions to shrink trivial effects toward zero. Ridge regression (Hoerl and Kennard 1970) is a shrinkage procedure and can be obtained using independent and identical normal priors centered at zero, with the degree of shrinkage controlled by the prior variance. The least absolute shrinkage and selection operator (LASSO) is another shrinkage method that has been widely used in regression analysis for large models (Tibshirani 1996). It minimizes the residual sum of squares constraining the sum of absolute values of the regression coefficients, 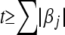 for
for 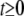 , if the response and the predictors are standardized. This special constraint allows some estimated regression coefficients to be exactly zero. This selective or individualized shrinkage allows LASSO to handle extremely large models. Mathematically, the LASSO estimates of regression coefficients can be achieved by
, if the response and the predictors are standardized. This special constraint allows some estimated regression coefficients to be exactly zero. This selective or individualized shrinkage allows LASSO to handle extremely large models. Mathematically, the LASSO estimates of regression coefficients can be achieved by
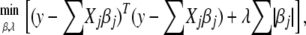 |
where 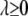 is a Lagrange multiplier, which relates implicitly to the bound t and controls the degree of shrinkage. The LASSO estimates of coefficients can be efficiently computed via the LARS algorithm of Efron et al. (2004). The LASSO procedure can be interpreted as a Bayesian posterior mode estimate when assigning an independent double-exponential prior to each
is a Lagrange multiplier, which relates implicitly to the bound t and controls the degree of shrinkage. The LASSO estimates of coefficients can be efficiently computed via the LARS algorithm of Efron et al. (2004). The LASSO procedure can be interpreted as a Bayesian posterior mode estimate when assigning an independent double-exponential prior to each 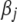 (e.g., Tibshirani 1996; Yuan and Lin 2005; Park and Casella 2007). Recently, Xu (2007) successfully applied the LASSO method to estimate the epistatic effects of QTL. However, the LASSO provides no estimate for the residual error variance and no interval estimate for a regression coefficient and needs to predetermine the parameter
(e.g., Tibshirani 1996; Yuan and Lin 2005; Park and Casella 2007). Recently, Xu (2007) successfully applied the LASSO method to estimate the epistatic effects of QTL. However, the LASSO provides no estimate for the residual error variance and no interval estimate for a regression coefficient and needs to predetermine the parameter 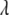 (Tibshirani 1996; Efron et al. 2004).
(Tibshirani 1996; Efron et al. 2004).
There are some other shrinkage methods that have been applied to mapping multiple QTL. The method of Xu (2003) simultaneously fits maker effects of the entire genome in a regression model and assigns each effect a normal prior with mean 0 and an effect-specific variance (also see Wang et al. 2005; Hoti and Sillanpää 2006; Huang et al. 2007). The variance parameters are further assigned the noninformative Jeffrey's prior, which in turn induces improper priors on the coefficients. Meuwissen et al. (2001) and Xu (2007) used scaled inverse-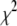 distributions with predetermined hyperparameters as priors for the effect-specific variance parameters. Rather than presetting the hyperparameters in the priors that control the degree of shrinkage, it would be appealing to treat the hyperparameters as unknowns and estimate them from the data so that the model can shrink the coefficients as much as can be justified by the data. Park and Casella (2007) set up a hyperprior for the hyperparameter in the LASSO model and developed a Gibbs sampler to fit the hierarchical model. R. B. O'Hara and M. J. Sillanpää (unpublished results) conducted a comparison with several Bayesian model selection and shrinkage methods, including Bayesian LASSO and the method of Xu (2003).
distributions with predetermined hyperparameters as priors for the effect-specific variance parameters. Rather than presetting the hyperparameters in the priors that control the degree of shrinkage, it would be appealing to treat the hyperparameters as unknowns and estimate them from the data so that the model can shrink the coefficients as much as can be justified by the data. Park and Casella (2007) set up a hyperprior for the hyperparameter in the LASSO model and developed a Gibbs sampler to fit the hierarchical model. R. B. O'Hara and M. J. Sillanpää (unpublished results) conducted a comparison with several Bayesian model selection and shrinkage methods, including Bayesian LASSO and the method of Xu (2003).
In this article, we propose several Bayesian hierarchical models for mapping multiple QTL that simultaneously fit and estimate all possible genetic effects associated with all markers across the entire genome. We use prior distributions for the genetic effects that are scale mixtures of normal distributions with mean zero and unknown effect-specific variances. We consider two types of priors for the variances, exponential and scaled inverse-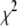 distributions, which result in a Bayesian version of the LASSO model and the well-known Student's t model, respectively. We treat all hyperparameters as unknowns and estimate them along with other parameters. We fit the models in a fully Bayesian approach, employing the Markov chain Monte Carlo (MCMC) simulation to generate posterior samples from the joint posterior distribution. Our methods give not only point estimates but also interval estimates of all parameters and provide natural means of assessing model uncertainty.
distributions, which result in a Bayesian version of the LASSO model and the well-known Student's t model, respectively. We treat all hyperparameters as unknowns and estimate them along with other parameters. We fit the models in a fully Bayesian approach, employing the Markov chain Monte Carlo (MCMC) simulation to generate posterior samples from the joint posterior distribution. Our methods give not only point estimates but also interval estimates of all parameters and provide natural means of assessing model uncertainty.
MULTIPLE-QTL MODEL
We describe our method primarily for a mapping population with only two segregating genotypes, e.g., a backcross, double-haploid (DH), or recombinant inbred line (RIL). The method can be extended to other types of population. Assume that we observe many markers across the genome. The aim of QTL mapping is to identify which markers are tightly linked to genes with detectable effects and to estimate the magnitudes of the effects. For a continuously distributed trait, the observed phenotypic value yi of individual i can be described by the linear regression model
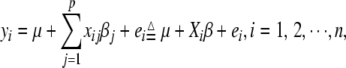 |
(1) |
where p is the number of markers,  is the overall mean,
is the overall mean, 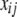 denotes the genotype of marker j for individual i and is defined as −0.5 and 0.5 for the two genotypes in the mapping population, the coefficient
denotes the genotype of marker j for individual i and is defined as −0.5 and 0.5 for the two genotypes in the mapping population, the coefficient 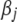 represents the main effect of marker j,
represents the main effect of marker j, 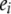 is the residual error assumed to follow a
is the residual error assumed to follow a 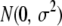 distribution,
distribution, 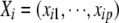 , and
, and 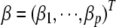 .
.
In practice, some marker genotypes may be missing. There are two commonly used methods to deal with missing marker data. The first approach takes the uncertainty in missing genotypes into account by treating the missing genotypes as unknowns and sampling them in the MCMC update procedure. The second approach is similar to the regression model of Haley and Knott (1992) and replaces all missing genotypes by their expected values conditioning on the observed marker data, using the multipoint method (Jiang and Zeng 1997). Although the second method ignores the uncertainty of the estimated marker genotypes, it has a big computational advantage over the first method. Both approaches can be incorporated into our Bayesian method. For simplicity and computational convenience, this study considers only the second method.
PRIOR DISTRIBUTIONS
Data are usually sufficient to estimate the overall mean 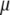 and the residual variance
and the residual variance 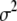 . Thus, we can use any reasonable noninformative prior distributions for these parameters. For example, we can use an independent, flat prior for
. Thus, we can use any reasonable noninformative prior distributions for these parameters. For example, we can use an independent, flat prior for  , i.e.,
, i.e.,  , and a noninformative scale-invariant prior 1/
, and a noninformative scale-invariant prior 1/ for
for 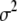 .
.
QTL data typically contain a large number of markers, leading to many coefficients in model (1). However, most of the coefficients are expected to have no or only weak effects on the phenotype. Only a few coefficients may have notable effects (see, e.g., Xu 2003). To incorporate this prior knowledge into our analysis, therefore, we set up a prior distribution that gives each coefficient 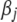 a high probability of being near zero and in the meantime gives each coefficient a chance to take a large effect.
a high probability of being near zero and in the meantime gives each coefficient a chance to take a large effect.
A commonly used prior that has the above properties is the double exponential (also called Laplace) distribution
 |
(2) |
(Tibshirani 1996; Park and Casella 2007), where 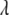 is a hyperparameter. With the double-exponential prior, the posterior mode estimate of the coefficients
is a hyperparameter. With the double-exponential prior, the posterior mode estimate of the coefficients  is the LASSO estimate (Tibshirani 1996; Park and Casella 2007). Another wide-tailed distribution is the well-known Student's t distribution,
is the LASSO estimate (Tibshirani 1996; Park and Casella 2007). Another wide-tailed distribution is the well-known Student's t distribution,
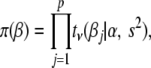 |
(3) |
where the hyperparameters 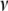 ,
, 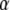 , and
, and 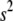 are the degrees of freedom, the location, and the scale parameters, respectively (see, e.g., Sorensen and Gianola 2002; Gelman et al. 2003; Varona et al. 2005).
are the degrees of freedom, the location, and the scale parameters, respectively (see, e.g., Sorensen and Gianola 2002; Gelman et al. 2003; Varona et al. 2005).
Both the double-exponential distribution and Student's t distribution can be presented as a two-level hierarchical model (see, e.g., Andrews and Mallows 1974; Griffin and Brown 2006). Compared with their original forms, the two-level hierarchical models are easier to deal with both analytically and computationally. In the two-level hierarchical models, the first level assumes that the coefficients  follow independent normal distributions with mean zero and unknown variances
follow independent normal distributions with mean zero and unknown variances 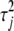 ,
,
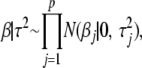 |
(4) |
where 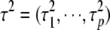 . At the second level, we assume that the variances
. At the second level, we assume that the variances  follow some specified independent prior distributions
follow some specified independent prior distributions
 |
(5) |
where 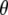 includes all the hyperparameters. The above two-level priors result in a scale mixture of normal distributions for the coefficients
includes all the hyperparameters. The above two-level priors result in a scale mixture of normal distributions for the coefficients 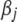 ,
,
 |
(6) |
where 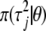 is referred to as the mixing distribution. From this, we see that two-level hierarchical models introduce different variance parameters for different coefficients and hence induce different amounts of shrinkage in the estimates of different coefficients. The variance components are not the parameters of interest, but they are useful intermediate quantities to facilitate better inferences for the individual regression coefficients (e.g., Gelman 2005).
is referred to as the mixing distribution. From this, we see that two-level hierarchical models introduce different variance parameters for different coefficients and hence induce different amounts of shrinkage in the estimates of different coefficients. The variance components are not the parameters of interest, but they are useful intermediate quantities to facilitate better inferences for the individual regression coefficients (e.g., Gelman 2005).
The double-exponential distribution (2) can be presented as a mixture of normal distributions (4) with the mixing distribution 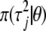 being an exponential distribution Expon(
being an exponential distribution Expon( ) or equivalently a gamma distribution Gamma(1,
) or equivalently a gamma distribution Gamma(1, 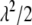 ),
),
 |
(7) |
where 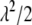 is the inverse scale that needs to be carefully preset or estimated from the data.
is the inverse scale that needs to be carefully preset or estimated from the data.
For any values of 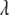 , the mode of the exponential distribution Expon(
, the mode of the exponential distribution Expon( ) = 0, meaning that the “most likely” values of
) = 0, meaning that the “most likely” values of 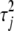 and thus
and thus 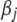 are zero. However, the variance of
are zero. However, the variance of 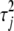 is
is 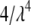 , strongly depending on the value of
, strongly depending on the value of  . Instead of presetting a value for
. Instead of presetting a value for 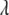 , it is appealing to assign a prior distribution to
, it is appealing to assign a prior distribution to 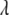 also so that
also so that 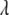 can be estimated along with other parameters. This procedure obviates the choice of
can be estimated along with other parameters. This procedure obviates the choice of 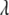 and automatically accounts for the uncertainty in its selection that affects the estimates of regression coefficients (Park and Casella 2007). For convenience, we regard
and automatically accounts for the uncertainty in its selection that affects the estimates of regression coefficients (Park and Casella 2007). For convenience, we regard 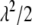 , instead of
, instead of 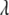 , as the parameter of interest. Following Park and Casella (2007), we assign a conjugate gamma prior Gamma(a, b), a > 0, b > 0, to the parameter
, as the parameter of interest. Following Park and Casella (2007), we assign a conjugate gamma prior Gamma(a, b), a > 0, b > 0, to the parameter 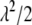 . Assigning a prior distribution to
. Assigning a prior distribution to 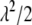 provides a way to estimate
provides a way to estimate 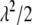 . We set a and b as small values (e.g., a = 0.1 and b = 0.1) so that the prior for
. We set a and b as small values (e.g., a = 0.1 and b = 0.1) so that the prior for  is essentially noninformative.
is essentially noninformative.
Student's t distribution (3) can be expressed as a mixture of normal distributions (4) with the mixing distribution  being a conjugate scaled inverse-
being a conjugate scaled inverse-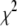 distribution
distribution 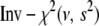 or equivalently an inverse gamma distribution Inv-gamma(
or equivalently an inverse gamma distribution Inv-gamma(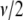 ,
,  ),
),
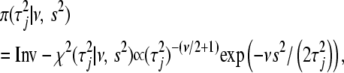 |
(8) |
where the hyperparameters 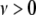 and
and  represent the degrees of freedom and the scale of the distribution, respectively.
represent the degrees of freedom and the scale of the distribution, respectively.
The amount of shrinkage in the prior (8) depends on the values of the hyperparameters  and
and 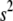 . The improper distribution
. The improper distribution  , which is equivalent to the uniform distribution on
, which is equivalent to the uniform distribution on 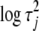 , has the form of the inverse-
, has the form of the inverse-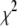 density with
density with 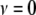 and
and  and can be taken as a limit of the proper inverse-
and can be taken as a limit of the proper inverse-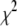 or inverse-gamma densities. This improper prior in turn induces an improper prior for
or inverse-gamma densities. This improper prior in turn induces an improper prior for  of the form
of the form  (Griffin and Brown 2006). In the class of inverse-
(Griffin and Brown 2006). In the class of inverse- densities,
densities, 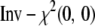 has the constant degree of shrinkage in the coefficient estimates. Xu (2003) actually used this improper prior
has the constant degree of shrinkage in the coefficient estimates. Xu (2003) actually used this improper prior 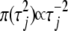 in his shrinkage QTL mapping.
in his shrinkage QTL mapping.
We here treat 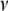 and
and 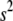 as unknown parameters, obviating the choice of (
as unknown parameters, obviating the choice of ( ,
, 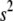 ) and automatically accounting for the uncertainty in its selection that affects the estimates of the regression coefficients. We assign a uniform density on 1/
) and automatically accounting for the uncertainty in its selection that affects the estimates of the regression coefficients. We assign a uniform density on 1/ for the range (0, 1] and a uniform distribution on
for the range (0, 1] and a uniform distribution on  for the range (0, A] with A being a large number (see Gelman et al. 2003).
for the range (0, A] with A being a large number (see Gelman et al. 2003).
The prior (4) on the coefficients  is independent of the residual variance and has been commonly used in the literature. Park and Casella (2007) recently proposed an alternative version of the prior on
is independent of the residual variance and has been commonly used in the literature. Park and Casella (2007) recently proposed an alternative version of the prior on 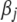 that is dependent on the residual variance
that is dependent on the residual variance 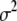 ,
,
 |
(9) |
where 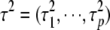 . At the second level, the prior distributions on
. At the second level, the prior distributions on  take the same form as (7) or (8). This conditional prior may have some advantages; for example, it results in that
take the same form as (7) or (8). This conditional prior may have some advantages; for example, it results in that  is unitless and posteriors induced from (9) generally do not have more than one mode (Park and Casella 2007).
is unitless and posteriors induced from (9) generally do not have more than one mode (Park and Casella 2007).
We now have introduced two different priors for 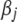 and two different priors for
and two different priors for 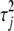 . The two priors for
. The two priors for 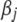 are
are  and
and  , respectively. The two priors for
, respectively. The two priors for  are
are  and
and  , respectively. This yields four possible combinations and thus four different models. These four models are called model I with prior
, respectively. This yields four possible combinations and thus four different models. These four models are called model I with prior 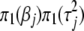 , model II with prior
, model II with prior  , model III with prior
, model III with prior  , and model IV with prior
, and model IV with prior 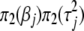 .
.
POSTERIOR COMPUTATION
We fit the models using the MCMC algorithm, applied to the joint posterior distribution of all the parameters  . The joint posterior distribution can be expressed as
. The joint posterior distribution can be expressed as
 |
(10) |
where  ,
,  ,
,  ,
,  represents
represents  for models I and II and (
for models I and II and ( ) for models III and IV,
) for models III and IV,  is the normal density function
is the normal density function  , and other terms in the right-hand side are the priors defined in the last section. For notational convenience, we suppress the dependence on X here and afterward.
, and other terms in the right-hand side are the priors defined in the last section. For notational convenience, we suppress the dependence on X here and afterward.
With the two-level hierarchical models as set up in the last section, the joint posterior distribution  can be simulated using the MCMC algorithm, alternately updating
can be simulated using the MCMC algorithm, alternately updating  from the normal distribution
from the normal distribution  , updating each
, updating each  from the normal distribution
from the normal distribution  , where
, where  represents all elements of
represents all elements of  except
except  , updating
, updating 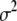 from the inverse-
from the inverse- distribution
distribution  , updating each
, updating each  from the inverse Gaussian (for models I and II) or inverse-
from the inverse Gaussian (for models I and II) or inverse- distribution (for models III and IV), and updating
distribution (for models III and IV), and updating  from the gamma distribution
from the gamma distribution  (for models I and II), or updating
(for models I and II), or updating 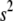 from the gamma distribution
from the gamma distribution  and updating
and updating 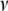 from
from  , using a Metropolis step (for models III and IV).
, using a Metropolis step (for models III and IV).
The two-level hierarchical modeling allows us to easily derive the above conditional posterior distributions (see appendix a). For all the four proposed models, however, the above MCMC algorithms can be performed directly using the Bayesian software WinBUGS14 (Spiegelhalter et al. 2002), and thus we actually do not need to derive the conditional posteriors. However, the expressions of the conditional posteriors allow us to develop faster, purpose-built programs. The WinBUGS code implementing these four hierarchical models can be obtained by request from the first author.
The MCMC algorithm proceeds to draw each unknown from its fully conditional posterior distribution, given the current values of all other unknowns and the observed data. Each iteration of the MCMC algorithm cycles through all elements of  . This process continues for a large number of iterations to obtain random samples from the joint posterior distribution. We run multiple parallel chains with overdispersed starting points and use the potential scale reduction factor
. This process continues for a large number of iterations to obtain random samples from the joint posterior distribution. We run multiple parallel chains with overdispersed starting points and use the potential scale reduction factor  that compares the between- and within-sequence variances for any scalar estimands, to monitor convergence (Gelman et al. 2003). We use the condition of
that compares the between- and within-sequence variances for any scalar estimands, to monitor convergence (Gelman et al. 2003). We use the condition of  < 1.1 for all scalar estimands of interest as the criteria of convergence. Once our simulations have converged, we continue to draw thousands of simulations and treat them as a sample from the joint posterior distribution. We thin the sequences by keeping every kth simulation draw from each sequence and discarding the rest (e.g., k = 100).
< 1.1 for all scalar estimands of interest as the criteria of convergence. Once our simulations have converged, we continue to draw thousands of simulations and treat them as a sample from the joint posterior distribution. We thin the sequences by keeping every kth simulation draw from each sequence and discarding the rest (e.g., k = 100).
POSTERIOR SUMMARY AND INTERPRETATION
The proposed Bayesian hierarchical models can provide various graphical and tabular summaries that assess the contribution of individual loci while adjusting for effects of all other possible loci. One of such summaries is the estimate of the genetic effects  associated with each marker. Furthermore, using the posterior samples of
associated with each marker. Furthermore, using the posterior samples of  and the observed values
and the observed values  , we can also calculate the proportion of the phenotypic variance explained by each effect (i.e., heritability),
, we can also calculate the proportion of the phenotypic variance explained by each effect (i.e., heritability),  , where Vy is the phenotypic variance and
, where Vy is the phenotypic variance and  is the sample variance of
is the sample variance of  . The heritability
. The heritability  is independent of the scale of the phenotype, while the effect size
is independent of the scale of the phenotype, while the effect size  depends on it. The fully Bayesian analysis usually uses the posterior median or mean to estimate
depends on it. The fully Bayesian analysis usually uses the posterior median or mean to estimate  and
and  and also can provide the posterior interval for the estimate.
and also can provide the posterior interval for the estimate.
The Bayesian hierarchical models shrink the “insignificant” coefficients to zero, and hence the posterior estimates of 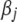 and
and  can guide variable selection. However, these posterior summaries do not automatically provide a criterion to declare that an effect is “in” the model. Hoti and Sillanpää (2006) suggest setting a threshold value, c, such that the standardized effect
can guide variable selection. However, these posterior summaries do not automatically provide a criterion to declare that an effect is “in” the model. Hoti and Sillanpää (2006) suggest setting a threshold value, c, such that the standardized effect  is included if
is included if  . Note that the summary statistic, heritability, is essentially the same as the standardized effects in Hoti and Sillanpää (2006). Yang and Xu (2007) proposed using a Wald test statistic.
. Note that the summary statistic, heritability, is essentially the same as the standardized effects in Hoti and Sillanpää (2006). Yang and Xu (2007) proposed using a Wald test statistic.
We here construct an alternative measurement that is similar to the widely used LOD score in the traditional QTL mapping approach if the phenotype is affected by a single QTL (Lander and Botstein 1989), following the idea of Yandell et al. (2007) (also see the vignette in the package R/qtlbim). We call this statistic the approximate Bayesian LOD score. The approximate Bayesian LOD score can be conveniently compared with the LOD curve obtained from the traditional interval-mapping approach. Our Bayesian LOD score considers the contribution of a given locus to the LOD after adjusting effects for all other possible loci. For marker j, the Bayesian LOD score (BLOD) is calculated as
 |
(11) |
where the values of  ,
,  , and
, and  are the simulated draws in a certain iteration, and
are the simulated draws in a certain iteration, and  , the mean of the conditional posterior
, the mean of the conditional posterior  . Note that although we adjust for the residual error variance, we use the simulated draws under the full model (including
. Note that although we adjust for the residual error variance, we use the simulated draws under the full model (including  ) to calculate the likelihood function for the reduced model (without
) to calculate the likelihood function for the reduced model (without  ).
).
REAL DATA ANALYSIS
The well-known barley data set from the North American Barley Genome Mapping Project (Tinker et al. 1996) was analyzed using the four proposed Bayesian hierarchical models. This data set has been extensively analyzed using different methods (Tinker et al. 1996; Xu 2003, 2007; Yi et al. 2003, 2005; Zhang et al. 2005; Xu and Jia 2007). The data were collected from a doubled-haploid population that contained n = 145 lines; each was grown in 25 different environments. The phenotype analyzed was the average value of kernel weight across environments. A total of p = 127 mapped markers covering a genome of 1500 cM (seven chromosomes) were used in the analysis. Observed marker genotypes were coded as −0.5 for one genotype and +0.5 for the other genotype. The data contain ∼5% missing marker genotypes. The missing marker genotypes were replaced by their expected values conditioning on the observed marker data using the multipoint method. We simultaneously modeled all 127 marker effects in the linear regression (1).
For all four proposed models, the MCMC simulations were performed in the Bayesian software WinBUGS14 (Spiegelhalter et al. 2002), linked from the R statistical computing software (Sturtz et al. 2005; R Development Core Team 2006), which we can use to conveniently plot the results. For each analysis, we ran three parallel simulation sequences with starting points randomly generated from the prior distributions. The models with the dependent prior (9) (models III and IV) were found to converge slightly quicker than those with the independent prior (4) (models I and II). For all four models, the potential scale reduction factor  values fell below 1.1 for all parameters after ∼5000 iterations, indicating that the chains converged adequately (data not shown). For each of the three parallel sequences, therefore, we ran a total of 20,000 iterations and discarded the first halves to allow adequate convergence. The remaining 10,000 posterior samples of each sequence were thinned to 1000 for estimating the posterior distribution of quantities of interest by keeping every 10th simulation draw from each sequence and discarding the rest. On a dual processor 2.4-GHz machine, each analysis took ∼4.5 hr.
values fell below 1.1 for all parameters after ∼5000 iterations, indicating that the chains converged adequately (data not shown). For each of the three parallel sequences, therefore, we ran a total of 20,000 iterations and discarded the first halves to allow adequate convergence. The remaining 10,000 posterior samples of each sequence were thinned to 1000 for estimating the posterior distribution of quantities of interest by keeping every 10th simulation draw from each sequence and discarding the rest. On a dual processor 2.4-GHz machine, each analysis took ∼4.5 hr.
To investigate whether or not posterior inferences are affected by the values of the shape a and the inverse scale b in the gamma prior Gamma(a, b), we analyzed models I and II with four different values of (a, b), a = b = 0.01, 0.1, 0.5, 1. Under the four analyses, the posterior estimates of the parameters  were identical, but the inverse scale
were identical, but the inverse scale  differed slightly.
differed slightly.
Figure 1 shows the posterior medians and the 95% posterior intervals for the marker effects  and the proportion of the phenotypic variance explained by each effect (i.e., heritability)
and the proportion of the phenotypic variance explained by each effect (i.e., heritability)  . Models I and II produced almost identical results, both being able to identify markers with large effects. The analyses showed four chromosomes (7, 1, 3, and 2) with strong evidence of QTL. The QTL on chromosome 3 was found to have an opposite effect on the phenotype to the other detected QTL and was missed in the analysis of Xu (2003), who used the improper prior
. Models I and II produced almost identical results, both being able to identify markers with large effects. The analyses showed four chromosomes (7, 1, 3, and 2) with strong evidence of QTL. The QTL on chromosome 3 was found to have an opposite effect on the phenotype to the other detected QTL and was missed in the analysis of Xu (2003), who used the improper prior  . The difference may also be caused by the different ways of treating missing marker genotypes for the two methods. As can be seen in Figure 1, the plots of the heritability can provide a clearer picture than those of the effect.
. The difference may also be caused by the different ways of treating missing marker genotypes for the two methods. As can be seen in Figure 1, the plots of the heritability can provide a clearer picture than those of the effect.
Figure 1.—

Posterior medians (points) and 95% intervals (shaded lines) for genetic effects and the proportion of the phenotypic variance explained by each effect (i.e., heritability). (Top) Plots drawn from model I; (bottom) plots for model II. Inner tick marks on the x-axis represent the marker positions.
Figure 2 shows the histograms of the inverse scale  for the analyses of models I and II with a = b = 0.1. We can see that the hyperparameter
for the analyses of models I and II with a = b = 0.1. We can see that the hyperparameter  can be estimated with quite high precision. For model I, the inverse scale parameter was estimated at 32.0, with a 95% posterior interval of [20.6, 50.5]. However, model II provided a much smaller estimate, with a posterior median of 5.1 and a 95% posterior interval of [2.8, 9.8]. We know that in the exponential distribution a larger inverse scale forces stronger shrinkage. Figure 1 shows that models I and II perform at the same level of shrinkage in the coefficient estimates. This can also be shown by looking at the estimated
can be estimated with quite high precision. For model I, the inverse scale parameter was estimated at 32.0, with a 95% posterior interval of [20.6, 50.5]. However, model II provided a much smaller estimate, with a posterior median of 5.1 and a 95% posterior interval of [2.8, 9.8]. We know that in the exponential distribution a larger inverse scale forces stronger shrinkage. Figure 1 shows that models I and II perform at the same level of shrinkage in the coefficient estimates. This can also be shown by looking at the estimated  , which was 0.3, <1.
, which was 0.3, <1.
Figure 2.—

Histogram of the posterior samples for the inverse scale of the exponential prior on the variances. The dotted lines represent the posterior 5, 50, and 95% quantiles. The left and right plots show inferences for models I and II, respectively.
Figure 3 depicts the posterior medians and the 95% posterior intervals for marker effect  and the heritability
and the heritability  from the analyses of models III and IV. Models III and IV produced identical results, similar to those from models I and II. However, the Inv-
from the analyses of models III and IV. Models III and IV produced identical results, similar to those from models I and II. However, the Inv- prior distributions on
prior distributions on  produced slightly larger posterior medians and wider posterior intervals for
produced slightly larger posterior medians and wider posterior intervals for 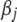 and
and  . The degrees of freedom and scale parameters can be estimated from the data. Figure 4 shows the histograms of these two hyperparameters for the analyses of models III and IV. The two models produced similar estimates for the degrees of freedom, with the posterior median at 2.1 and the 95% posterior interval of [1.2, 5.3]. However, the estimate of the scale parameter for model IV was three times as large as that for model III.
. The degrees of freedom and scale parameters can be estimated from the data. Figure 4 shows the histograms of these two hyperparameters for the analyses of models III and IV. The two models produced similar estimates for the degrees of freedom, with the posterior median at 2.1 and the 95% posterior interval of [1.2, 5.3]. However, the estimate of the scale parameter for model IV was three times as large as that for model III.
Figure 3.—

Posterior medians (points) and 95% intervals (shaded lines) for genetic effects and the proportion of the phenotypic variance explained by each effect (i.e., heritability). (Top) Plots drawn from model III; (bottom) plots for model IV. Inner tick marks on the x-axis represent the marker positions.
Figure 4.—

Histogram of the posterior samples for the degrees of freedom and scale of the Inv- prior on variances. The dotted lines represent the posterior 5, 50, and 95% quantiles. The top and bottom plots show inferences for models III and IV, respectively.
prior on variances. The dotted lines represent the posterior 5, 50, and 95% quantiles. The top and bottom plots show inferences for models III and IV, respectively.
Figure 5 (top) shows the posterior medians and the 95% posterior intervals for the Bayesian LOD score for model I. The other three models produced similar estimates (data not shown). For comparison, Figure 5 (bottom) also shows the LOD score curve calculated by the traditional interval mapping based on a single-QTL model. If we use the standard threshold value of 3.2, the interval mapping identified only two regions (on chromosomes 7 and 1, respectively), which have larger effects in our analyses. Our Bayesian models produced much higher estimates of LOD score and detected more significant QTL.
Figure 5.—

The top plot shows posterior medians (points) and 95% intervals (shaded lines) for the approximate Bayesian LOD score from model I, and the bottom plot shows the LOD score curve from traditional interval mapping. The dotted lines represent the standard threshold value of 3.2. Inner tick marks on the x-axis represent the marker positions.
In the proposed two-level hierarchical models, the amount of shrinkage in the coefficient estimates is largely determined by the hyperparameters in the priors. It is expected that optimal values of the hyperparameters depend on the data and the number of variables included, which makes it difficult to preset their values. To illustrate this, we analyzed the data with only markers on chromosomes 1, 3, and 7 included in the model. Figure 6 displays the plots of marker effects, showing that we were still able to detect the same QTL as in our previous analyses. As shown in Figure 7, however, the posterior estimates of the hyperparameters were different from those in the previous analyses.
Figure 6.—

Posterior medians (points) and 95% intervals (shaded lines) for genetic effects from all four models using markers on chromosomes 1, 3, and 7. Inner tick marks on the x-axis represent the marker positions.
Figure 7.—

Histogram of the posterior samples for the hyperparameters of priors on variances. The dotted lines represent the posterior 5, 50, and 95% quantiles. The top plots show inferences for models I and II, respectively, the middle plots show inferences from model III, and the bottom plots show inferences from model IV.
DISCUSSION
We have presented four Bayesian hierarchical models that can be used to simultaneously fit and estimate all possible genetic effects associated with all molecular markers across the entire genome. We found that these four methods perform equally well. It is known a priori that most markers have no or little effect on the phenotype, but if a marker has an effect, it could be large. To incorporate this prior knowledge into our analysis, we set up a continuous form of prior distribution for genetic effects that gives each effect a high probability of being near zero and also enables us to accommodate large effects. We have considered two such priors—double exponential distribution and Student's t distribution—that can be expressed as scale mixtures of normal distributions with mean zero and unknown variances distributed as exponential and scaled inverse- distributions, respectively. The former prior results in a Bayesian LASSO model (Tibshirani 1996; Yuan and Lin 2005; Park and Casella 2007), and the latter includes Jeffrey's noninformative prior as a special case (Gelman 2006).
distributions, respectively. The former prior results in a Bayesian LASSO model (Tibshirani 1996; Yuan and Lin 2005; Park and Casella 2007), and the latter includes Jeffrey's noninformative prior as a special case (Gelman 2006).
Both the exponential and the scaled inverse- distributions include hyperparameters that determine the degrees of shrinkage in the estimates of the variances and thus the genetic effects. A common practice is to preset values for the hyperparameters using methods like cross validation (Tibshirani 1996; Meuwissen et al. 2001; Xu 2003; Bao and Mallick 2004; Xu 2007). However, optimal values depend on the data and thus may be difficult to choose. A novel aspect of our approach is to treat all hyperparameters as unknowns and estimate them along with other parameters. This procedure allows us to control the amount of shrinkage by taking advantage of the characteristics of the data. In contrast, Jeffrey's prior includes no hyperparameter but always forces a constant degree of shrinkage. Xu (2003) showed that Jeffrey's prior yields good performance, but we observed that it converges more slowly than our proposed methods.
distributions include hyperparameters that determine the degrees of shrinkage in the estimates of the variances and thus the genetic effects. A common practice is to preset values for the hyperparameters using methods like cross validation (Tibshirani 1996; Meuwissen et al. 2001; Xu 2003; Bao and Mallick 2004; Xu 2007). However, optimal values depend on the data and thus may be difficult to choose. A novel aspect of our approach is to treat all hyperparameters as unknowns and estimate them along with other parameters. This procedure allows us to control the amount of shrinkage by taking advantage of the characteristics of the data. In contrast, Jeffrey's prior includes no hyperparameter but always forces a constant degree of shrinkage. Xu (2003) showed that Jeffrey's prior yields good performance, but we observed that it converges more slowly than our proposed methods.
We fit the models in a fully Bayesian approach, employing the MCMC simulation to generate posterior samples from the joint posterior distribution, which can be used to make various posterior inferences. Although computationally intensive, it is easy to implement and provides not only point estimates but also interval estimates of all parameters. We have proposed graphical tools to summarize the posterior samples, notably the approximate Bayesian LOD score that can be easily compared with the results from traditional interval mapping. However, we realize that these tools still are informal and descriptive, due to the lack of formal threshold value to select markers. A formal choice of threshold value will be a topic of future research.
The fully Bayesian approach enables us to obtain much richer inferences about the models than most non-Bayesian analyses. In practice, however, it is desirable to have a quicker calculation that merely looks for posterior modes rather than fully investigating the posterior distribution. The original LASSO estimates for linear regression coefficients are equivalent to Bayesian posterior mode estimates when the coefficients have independent and identical double-exponential priors (Tibshirani 1996). The two-level hierarchical formulation enables us to find posterior modes using various algorithms, e.g., conditional maximization (Zhang and Xu 2005), the EM algorithm, and its extensions (Figueiredo 2003; Foster et al. 2007; Xu 2007). However, these methods have been developed on the basis of preset hyperparameters. Further investigation is necessary to extend the mode-searching algorithms to the proposed hierarchical models.
The proposed models have been used to fit all markers and estimate their effects. The methods can be easily extended to detect QTL within marker intervals. The idea is to insert loci within each marker interval as possible positions of QTL and put a reasonable number of loci on each chromosome with positions treated as parameters (Wang et al. 2005). An additional Metropolis step is then used to update the positions. Future research also will consider epistatic effects and extensions to complicated experimental crosses. Computationally efficient algorithms are an essential feature for the practical analysis of these more complicated cases. We are in the process of developing purpose-specific computer programs that would significantly reduce the computing time. Our hierarchical models assign marker effects independent priors with a single prior mean. However, it is desirable to develop hierarchical priors to allow shrinkage of coefficients toward multiple prior means with the locations of these means unknown and to accommodate the spatial covariance structure along markers (Gianola et al. 2003).
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants R01 GM069430, HL080812, and GM077490 to N.Y. and by the National Science Foundation grant DBI-0345205 and the National Research Initiative Plant Genome of the U.S. Department of Agriculture Cooperative State Research, Education, and Extension Service no. 2007-02784 to S.X.
APPENDIX A: CONDITIONAL POSTERIOR DISTRIBUTIONS
The two-level hierarchical models presented in this study have explicit forms of conditional posterior distributions for parameters and hyperparameters. These conditional posterior distributions are required to perform the MCMC algorithm. The posterior distributions are presented in this section.
Conditional on the parameters  , the models are the standard weighted linear regressions, and thus the conditional posterior distributions of
, the models are the standard weighted linear regressions, and thus the conditional posterior distributions of  are
are
 |
(A1) |
 |
(A2) |
 |
(A3) |
where  represents all elements of
represents all elements of  except
except  , and
, and  for models I and III or
for models I and III or  for models II and IV.
for models II and IV.
For models I and II, the conditional posterior distribution of  is inverse Gaussian,
is inverse Gaussian,
 |
(A4) |
A relatively simple algorithm is available for simulating from the inverse Gaussian distribution (Chhikara and Folks 1989). With the conjugate prior Gamma(a, b), the conditional posterior distribution of the hyperparameter 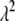 in the prior (7) is gamma
in the prior (7) is gamma
 |
(A5) |
For models III and IV, the conditional posterior distribution of  is inverse-
is inverse-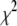 ,
,
 |
(A6) |
The conditional posterior distribution of the scale parameter  in the prior (8) is gamma
in the prior (8) is gamma
 |
(A7) |
The conditional posterior of the degrees of freedom  has no standard form. Therefore, a Metropolis step is required to update
has no standard form. Therefore, a Metropolis step is required to update  in each iteration.
in each iteration.
References
- Andrews, D. F., and C. L. Mallows, 1974. Scale mixtures of normal distributions. J. R. Stat. Soc. Ser. B 36 99–102. [Google Scholar]
- Bao, K., and B. K. Mallick, 2004. Gene selection using a two-level hierarchical Bayesian model. Bioinformatics 20 3423–3430. [DOI] [PubMed] [Google Scholar]
- Chhikara, R. S., and L. Folks, 1989. The Inverse Gaussian Distribution: Theory, Methodology, and Applications. Marcel Dekker, New York.
- Efron, B., T. Hastie, I. Johnstone and R. Tibshirani, 2004. Least angle regression. Ann. Stat. 32 407–499. [Google Scholar]
- Figueiredo, M. A. T., 2003. Adaptive sparseness for supervised learning. IEEE Trans. Patt. Anal. Machine Intell. 25 1150–1159. [Google Scholar]
- Foster, S. D., A. P. Verbyla and W. S. Pitchford, 2007. Incorporating LASSO effects into a mixed model for QTL detection. J. Agric. Biol. Environ. Stat. 12(2): 300–314. [Google Scholar]
- Gelman, A., 2005. Analysis of variance: why it is more important than ever (with discussion). Ann. Stat. 33 1–53. [Google Scholar]
- Gelman, A., 2006. Prior distributions for variance parameters in hierarchical models. Bayesian Anal. 1 515–533. [Google Scholar]
- Gelman, A., J. Carlin, H. Stern and D. Rubin, 2003. Bayesian Data Analysis. Chapman & Hall, London.
- Gianola, D., M. Perez-Enciso and M. A. Toro, 2003. On marker-assisted prediction of genetic value: beyond of the ridge. Genetics 163 347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, J. E., and P. J. Brown, 2006. Alternative prior distributions for variable selection with very many more variables than observations. Technical report. University of Warwick, Coventry, UK.
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69 315–324. [DOI] [PubMed] [Google Scholar]
- Hoerl, A. E., and R. W. Kennard, 1970. Ridge regression: biased estimation for nonorthogonal problems. Technometrics 12 55–67. [Google Scholar]
- Hoti, F., and M. J. Sillanpää, 2006. Bayesian mapping of genotype × expression interactions in quantitative and qualitative traits. Heredity 97 4–18. [DOI] [PubMed] [Google Scholar]
- Huang, H., C. D. Eversley, D. W. Threadgill and F. Zou, 2007. Bayesian multiple quantitative trait loci mapping for complex traits using markers of the entire genome. Genetics 176 2529–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, C., and Z.-B. Zeng, 1997. Mapping quantitative trait loci with dominant and missing markers in various crosses from two inbred lines. Genetica 101 47–58. [DOI] [PubMed] [Google Scholar]
- Kao, C. H., Z.-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, T., and G. Casella, 2007. The Bayesian Lasso. Technical report. University of Florida, Gainesville, FL.
- R Development Core Team, 2006. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org.
- Sillanpää, M. J., and E. Arjas, 1998. Bayesian mapping of multiple quantitative trait loci from incomplete inbred line cross data. Genetics 148 1373–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, D., and D. Gianola, 2002. Likelihood, Bayesian and MCMC Methods in Quantitative Genetics. Springer, New York.
- Spiegelhalter, D., A. Thomas, N. Best and D. Lunn, 2002. BUGS: Bayesian Inference Using Gibbs Sampling, Version 1.4. MRC Biostatistics Unit, Cambridge, UK. www.mrc-bsu.cam.ac.uk/bugs/.
- Sturtz, S., U. Ligges and A. Gelman, 2005. R2WinBUGS: a package for running WinBUGS from R. J. Stat. Software 12 1–16. [Google Scholar]
- Tibshirani, R., 1996. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B 58 267–288. [Google Scholar]
- Tinker, N. A., D. E. Mather, B. G. Rossnagel, K. J. Kasha and A. Kleinhofs, 1996. Regions of the genome that affect agronomic performance in two-row barley. Crop Sci. 36 1053–1062. [Google Scholar]
- Varona, L., W. Mekkawy, D. Gianola and A. Blasco, 2005. A whole genome analysis using robust asymmetric distributions. Genet. Res. 88 143–151. [DOI] [PubMed] [Google Scholar]
- Wang, H., Y. M. Zhang, X. Li, G. L. Masinde, S. Mohan et al., 2005. Bayesian shrinkage estimation of quantitative trait loci parameters. Genetics 170 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., 2007. An empirical Bayes method for estimating epistatic effects of quantitative trait loci. Biometrics 63 513–521. [DOI] [PubMed] [Google Scholar]
- Xu, S., and Z. Jia, 2007. Genome-wide analysis of epistatic effects for quantitative traits in barley. Genetics 175 1955–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell, B. S., T. Mehta, S. Banerjee, D. Shriner, R. Venkataraman et al., 2007. R/qtlbim: QTL with Bayesian interval mapping in experimental crosses. Bioinformatics 23 641–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, R., and S. Xu, 2007. Bayesian shrinkage analysis of quantitative trait loci for dynamic traits. Genetics 176 1169–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., 2004. A unified Markov chain Monte Carlo framework for mapping multiple quantitative trait loci. Genetics 167 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and D. Shriner, 2008. Advances in Bayesian multiple QTL mapping in experimental designs. Heredity 100 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., V. George and D. B. Allison, 2003. Stochastic search variable selection for identifying quantitative trait loci. Genetics 164 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., B. S. Yandell, G. A. Churchill, D. B. Allison, E. J. Eisen et al., 2005. Bayesian model selection for genome-wide epistatic QTL analysis. Genetics 170 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, M., and Y. Lin, 2005. Efficient empirical Bayes variable selection and estimation in linear models. J. Am. Stat. Assoc. 100 1215–1225. [Google Scholar]
- Zhang, Y.-M., and S. Xu, 2005. A penalized maximum likelihood method for estimating epistatic effects of QTL. Heredity 95 96–104. [DOI] [PubMed] [Google Scholar]
- Zhang, M., K. L. Montooth, M. T. Wells, A. G. Clark and D. Zhang, 2005. Mapping multiple quantitative trait loci by Bayesian classification. Genetics 169 2305–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]