




Abstract
Alzheimer's disease (AD) is characterized by accumulation and deposition of Aβ peptides in the brain. Aβ deposition in cerebrovessels occurs in many AD patients and results in cerebral amyloid angiopathy (AD/CAA). Since Aβ can be transported across blood–brain barrier (BBB), aberrant Aβ trafficking across BBB may contribute to Aβ accumulation in the brain and CAA development. Expression analyses of 273 BBB-related genes performed in this study showed that the drug transporter, ABCG2, was significantly upregulated in the brains of AD/CAA compared with age-matched controls. Increased ABCG2 expression was confirmed by Q-PCR, Western blot, and immunohistochemistry. Abcg2 was also increased in mouse AD models, Tg-SwDI and 3XTg. Aβ alone or in combination with hypoxia/ischemia failed to stimulate ABCG2 expression in BBB endothelial cells; however, conditioned media from Aβ-activated microglia strongly induced ABCG2 expression. ABCG2 protein in AD/CAA brains interacted and coimmunoprecipitated with Aβ. Overexpression of hABCG2 reduced drug uptake in cells; however, interaction of Aβ1–40 with ABCG2 impaired ABCG2-mediated drug efflux. The role of Abcg2 in Aβ transport at the BBB was investigated in Abcg2-null and wild-type mice after intravenous injection of Cy5.5-labeled Aβ1–40 or scrambled Aβ40–1. Optical imaging analyses of live animals and their brains showed that Abcg2-null mice accumulated significantly more Aβ in their brains than wild-type mice. The finding was confirmed by immunohistochemistry. These results suggest that ABCG2 may act as a gatekeeper at the BBB to prevent blood Aβ from entering into brain. ABCG2 upregulation may serve as a biomarker of CAA vascular pathology in AD patients.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease characterized by accumulation and deposition of Aβ peptides in the brain and cerebral vessels (Selkoe 2001; Selkoe and Schenk, 2003). Deposition of Aβ peptides in brain parenchyma forms senile plaques; whereas deposition of Aβ peptides in cerebral vessels results in cerebral amyloid angiopathy (CAA), which is found in 82–98% of AD patients (Jellinger, 2002; Weller and Nicoll, 2003; Kinnecom et al., 2007). Recent studies have shown that Aβ oligomers are toxic to neuronal cells and result in synaptic dysfunction (Oddo et al., 2003; Selkoe 2008). Accumulation and deposition of Aβ peptides in cerebral vasculature induce significant damage to cerebral endothelial cells and lead to chronic vascular inflammation, neurovascular uncoupling, and vascular insufficiency (Jellinger, 2002; Preston et al., 2003; Weller and Nicoll, 2003; Kinnecom et al., 2007). Aβ peptides can be transported bidirectionally across the blood–brain barrier (BBB) (Zlokovic, 2008). Low-density lipoprotein receptor-related protein-1 (LRP-1) is involved in efflux of Aβ peptides from the brain, whereas the receptor for advanced glycation endproducts (RAGE) is involved in shuttling Aβ from the circulation into the brain (Deane et al., 2004a,b, 2008; Zlokovic, 2008). Soluble form of LRP (sLRP) plays an important role in Aβ binding and clearance in plasma (Sagare et al., 2007; Deane et al., 2008a). ApoE and ApoJ are involved in Aβ clearance from the brain (Zlokovic, 1996; Bell et al., 2007; Deane et al., 2008b); ApoE2 and ApoE3 are more efficient than ApoE4 in mediating Aβ clearance from the brain (Deane et al., 2008b). It is believed that aberrant trafficking of Aβ peptides across the BBB may contribute to the accumulation of Aβ in the brain and the development of CAA pathology in AD patients (Zlokovic, 2008). Based on this model, the heavy burden of Aβ in AD brain results not only from increased production in the brain but also from decreased clearance from the brain and increased uptake from the circulation (Zlokovic, 2008). Recent evidence suggests that Aβ transport and clearance across the BBB determine the concentrations of Aβ peptides in the brain (Zlokovic, 2008). However, the routes and mechanisms of Aβ transport and trafficking across the BBB still remain largely uncharacterized.
By analyzing the expression of 273 BBB-related genes in the brain tissues (occipital cortices) of AD/CAA patients versus age-matched nondemented (ND) controls, we found that the BBB drug efflux transporter, ABCG2, was significantly upregulated in AD/CAA brain at both the mRNA and protein levels. ABCG2 is a 72 kDa transmembrane protein belonging to ATP-binding cassette (ABC) transporter family and is involved in multidrug resistance in some cancer cells/tissues (known as breast cancer resistant protein, i.e., BCRP) (Doyle and Ross, 2003; Zhang and Stanimirovic, 2005). ABCG2 is a “half-transporter” and forms functional homodimers that operate as a drug efflux transporter (Doyle and Ross, 2003; Zhang and Stanimirovic, 2005). We have previously cloned ABCG2 from primary human brain endothelial cells (HBEC) and demonstrated that ABCG2 is a drug efflux transporter at the BBB (Zhang et al., 2003). This study demonstrates that ABCG2 interacts with Aβ peptides and may act as a gatekeeper at the BBB to prevent blood Aβ1–40 peptides from entering into the brain.
Materials and Methods
Chemicals and reagents.
Anti-human β-actin antibody, protease inhibitor cocktail, dimethyl pyrocarbonate (DEPC), and Hoechst 33342 were purchased from Sigma. Anti-human ABCG2 antibodies were purchased from Alexis Biochemicals (BXP-21) and Santa Cruz Biotechnology. Western blotting stripping buffer was purchased from Pierce Biotechnology. ECL plus Western blotting detection kit, Cy5 and Cy3 dyes, and Cy5.5 labeling kit were purchased from Amersham/Pharmacia Biosciences. RNeasy kit, PCR column (QIAquick), and plasmid DNA purification kit were purchased from QIAGEN. DMEM, reverse transcriptase, TaqDNA polymerase, restriction enzymes, Trizol reagent, and Lipofectamine 2000 were purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from Hyclone Synthetic Aβ1–40 peptide and scrambled Aβ40–1 peptide were purchased from Biopeptides at 1 mg/vial.
Cell cultures.
Primary HBEC were generously provided by Dr. A. Prat (Montreal Neurological Institute, Montreal) and maintained as described previously (Zhang et al., 2003). Human embryonic kidney epithelial 293 cells (HEK293) were maintained in DMEM supplemented with 10% FBS; no coating was required on culture dishes and media were changed every second day. Lipofectamine 2000 (Invitrogen) was used to transfect HEK293 cells with a plasmid vector pTT5SH8Q2 carrying a full-length cDNA encoding human ABCG2 as per manufacturer's instructions. The expression of cloned hABCG2 in transfected cells was examined by Western blotting and immunocytochemistry as described previously (Zhang et al., 2003; Xiong et al., 2008) and below. The cells at 48 h after transfection expressed high levels of ABCG2 and were used for further experiments.
Primary human microglial cells were generated from postmortem brains and maintained as described previously (Lue et al., 2001a,b; Walker et al., 2001, 2006; Walker and Lue, 2005). The cells were treated with 2 or 5 μm aggregated Aβ1–42 or solvent for 24 h as described (Lue et al., 2001a,b; Walker et al., 2001, 2006). The conditioned media were harvested and centrifuged in a bench top Microfuge at 17,900 × g for 5 min at 4°C to remove cell debris. Supernatants were frozen at −80°C until use.
Human and mouse brain tissue samples and immunohistochemistry.
The use of human brain tissues in this work was approved by the Research Ethics Board of National Research Council of Canada (NRC–REB). The brain tissue samples of AD, AD with CAA (AD/CAA), and age-matched ND controls were obtained from the Brain and Body Donation Program at the Sun Health Research Institute (Sun City, AZ). The consent for participation in the Program was approved by the Sun Health Institutional Review Board (IRB). Brain samples (occipital cortices) of 13 AD patients with CAA pathology, 13 AD patients (without histopathological CAA finding), and 12 age-matched ND controls were used, respectively. The patients were examined and diagnosed by neurologists, and postmortem brain samples were examined and diagnosed by neuropathologists. The diagnosis of cerebral amyloid angiopathy pathology was made according to the presence of Aβ deposition in superficial cortical blood vessels (Olichney et al., 1996), cortical arterioles or capillaries.
The animal experimental protocols were approved by the Animal Care Committee of NRC–Institute for Biological Sciences (NRC–IBS). Mouse brain samples of transgenic Alzheimer's models, 3XTg (generously provided by Dr. Balu Chakravarthy at the NRC–IBS, Ottawa) and Tg-SwDI (generously provided by Dr. Cheryl Wellington, University of British Columbia, Vancouver, BC, Canada) and relevant wild-type (wt) mice were obtained. Abcg2 knock-out (KO) and wt mice were obtained from the Taconic Farms. The Abcg2 KO and wt animals were genotyped following the Taconic's protocol. The brain samples were obtained after in vivo optical imaging and embedded in OCT for frozen sections.
The brain tissues were frozen-sectioned at 10 μm thickness. Cerebral vessels were visualized by staining with the fluorescein-labeled lectin Ulex Europea Agglutinin I (UEA-I) or tomato lectin (green color) for human and mouse brains, respectively, as described previously (Zhang et al., 2003; Mojsilovic-Petrovic et al., 2004). Immunohistochemistry was performed using a mouse monoclonal anti-Aβ IgG (6E10) or a mouse monoclonal anti-ABCG2 IgG (BXP-21) antibodies as described (Zhang et al., 2003; Xiong et al., 2008). A goat anti-mouse secondary antibody conjugated with Alexa 568 (Invitrogen) was used to visualize Aβ deposits or ABCG2 in the brains (red color).
RNA extraction.
Total RNA was extracted from the brain tissues and cell cultures using Trizol reagent (Invitrogen) following the manufacturer's instructions. RNA was dissolved in dimethyl pyrocarbonate (DEPC)-treated Milli-Q dH2O, and 1 μg of RNA from each sample was resolved on a 1.0% formaldehyde agarose gel as described previously (Zhang et al., 2003), and the quality of the RNA samples were confirmed. The RNA samples used in microarray analyses were further cleaned by using an RNeasy kit (QIAGEN).
Development of a custom oligonucleotide microarray for blood–brain barrier related genes and microarray analysis.
An oligonucleotide glass slide microarray representing 273 human genes known to be related to BBB functions was developed at the NRC–IBS. The gene list was compiled through literature search, abstracts of BBB and cerebral vascular biology conferences, and GenBank database mining. To enhance the “discovery” capacity of the microarray, all known members of various transporter families were included, such as those encoding all 49 ABC transporters, solute carrier family (SLC) 1, SLC2 (GLUT), SLC5 (SGLT), SLC6 (GAT), SLC7 (CAT/LAT/y+ system), SLC11 (DMT), SLC15 (PEPT), SLC16 (MCT), SLC17 (NPT/VGLU), SLC18 (VMAT/VAChT), SLC21/SCLO (OATP), SLC22 (OCT/OAT), SLC28 (CNT), SLC29 (ENT), SLC38 (System A), RAGE, scavenger receptors, LRPs, and the genes encoding tight/adherens junctions, angiogenic factors, inflammatory cytokines, extracellular matrix, BBB-specific signaling/transcription factors, enzymes and glycoproteins (supplemental Table 1, available at www.jneurosci.org as supplemental material). The full list of the arrayed genes is found in supplemental Table 2, available at www.jneurosci.org as supplemental material. A number of internal and external controls, such as ACTB (β-actin), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), HIST2H2AA, TUBA3, HPRT1, and a set of SpotReport Alien controls (Stratagene) were arrayed on the slides for quality control.
Fifty-mer oligonucleotides for each of the 273 genes were designed and synthesized at 3 mmol quantity by MWG Biotech. The microarray was designed and printed with each gene in triplicate on epoxy-coated Schott Nexterion slides (SCHOTT North America) and tested at the NRC–IBS Microarray Facility. The quality of the printed microarray slides was validated by using SpotReport Alien controls (Stratagene). The printed microarray slides were used within 12 weeks.
RNA samples were isolated from AD/CAA and ND brain tissues using Trizol reagent following the manufacturer's instructions. Six ND samples of best quality were pooled together at equal amounts as a common reference for the microarray analysis. Six AD/CAA samples of best quality were also pooled at equal amounts. A total of four labeling and hybridization reactions were carried out. In each reaction, two sets of slides with a total of four slides (two slides per set) were used and dye swap was used to minimize variations. RNA (15 μg) from each pooled sample was used in a reverse-transcription (cDNA) reaction and the cDNA was indirectly labeled with Cy3 or Cy5 or vise versa, respectively, following a modified protocol of University Health Network, University of Toronto (Ontario Cancer Institute, Toronto, ON, Canada). Alien RNAs (Alien 1–8) (Stratagene) were prepared following the manufacturer's instructions and were included in the labeling reactions.
Before the reverse transcription (RT; cDNA) reaction, a mixture of 15 μg of RNA from ND or AD/CAA, 1 μl of Alien mix (Alien1–8), and 0.75 μg of Anc Oligo dT (Cortec) in DEPC water was heated at 70°C for 10 min and cooled down to room temperature and then to 4°C. The following was added to the reaction: 8 μl of 5× first strand buffer (Invitrogen), 4 μl of 0.1 m DTT, 3 μl of 20 mm dNTPs (Invitrogen), 3 μl of 2 mm dTTP (Invitrogen), 3 μl of 2 mm aminoallyl dUTP (AAdUTP) (Sigma), 1 μl of Rnasin (Promega) and 200 units of SuperScript II reverse transcriptase (Invitrogen) to a final volume of 40 μl. The reaction was incubated at 42°C for 2 h and then cooled down to 4°C. The sample was purified on a Montage column (Fisher Scientific) as follows: Milli-Q water (400 μl) was loaded onto the column and the column spun at 1000 × g for 9 min. Eight microliter of 10N NaOH was added to cDNA sample and heated to 65°C for 15 min. The sample was neutralized with 12 μl of 2 m HEPES (free acid). Milli-Q water was added to the sample to make a final volume of 400 μl. The mixture was loaded onto the column and the column was spun at 1000 × g for 15 min at room temperature. The cDNA (AAcDNA) was eluted from the column by adding 5 μl of Milli-Q water to center of the column, incubating at room temperature for 2 min, and spinning inversely at 1000 × g for 2 min at 4°C. For dye coupling reaction, 3 μl of 0.3M NaHCO3, pH 9.0, was mixed with 5 μl of AAcDNA and 2 μl of Cy3 or Cy5 dye. Cy3 and Cy5 were purchased from Amersham/Pharmacia Biosciences and prepared in DMSO following the manufacturer's instructions. The mixture was incubated in the dark at room temperature for 1 h with occasional gentle rocking. The labeled sample was purified on a QIAGEN PCR column (QIAquick) as per instructions. A small aliquot of Cy5-labeled sample was run on a 1% agarose gel to check the efficiency of dye incorporation. The quantity of Cy3-or Cy5-labeled cDNAs was determined by OD readings with a spectrophotometer. The labeled cDNA was precipitated with 0.3 m NaOAc, pH 5.5, 10 μg of glycogen and one volume cold isopropanol at 17,900 × g for 10 min at 4°C and resuspended in 2.5 μl of Milli-Q water.
Cy3-labeled ND sample was pooled with Cy5-labeled AD/CAA sample or vise versa. The pooled sample (5 μl) was mixed with 40 μl of hybridization buffer containing 40% deionized formamide, 5× Denhardt's solution, 5× SSC solution, 1 mm sodium pyrophosphate, 50 mm Tris-HCl, pH 7.4, and 0.1% SDS. The mixed hybridization solution (45 μl) was then added onto a microarray slide with Hybrislip (Sigma) and incubated in a humid chamber at 42°C for 3 d in the dark. The hybridized microarray slides were washed first with a buffer containing 2× SSC, 0.2% SDS and 0.1 mm DTT, second with a buffer containing 2× SSC and 0.1 mm DTT, and last with a buffer containing 0.2× SSC and 0.1 mm DTT for 10 min/wash at room temperature, respectively. The slides were spun at 500 × g for 5 min to remove excess moisture and then scanned with ScanArray 5000 (Perkin-Elmer). Spot intensity data were obtained using QuantArray (Perkin-Elmer). These data were preprocessed using software Normalizer 3.0 developed in house at NRC–IBS (Walker et al., 2004) to correct for dye bias and sorted by p value (t test p < 0.05) and linear fold change >1.33.
Real-time quantitative RT-PCR.
Reverse transcription (cDNA synthesis), real-time qRT-PCR, and semiquantitative RT-PCR were performed as described previously (Zhang et al., 2003). The expression of ABCG2, β-actin, or GAPDH was detected using the primers (Table 1) designed according to published sequences in the GenBank. cDNA synthesis and qRT-PCR were run on each brain RNA sample. Semiquantitative RT-PCR was run for RNA samples isolated from cultured cells, and qRT-PCR was repeated for positive samples if necessary. Student's t test was performed to analyze the expressional difference between ND and AD/CAA samples.
Table 1.
Primers for real-time qRT-PCR and semi-quantitative RT-PCR
| Genes | Protocol | Sense primer | Anti-sense primers |
|---|---|---|---|
| ABCG2 | RT-PCR | 5′-CTGTAGCAACACTTCTCATGAC-3′ | 5′- TTCAGGTAGGCAATTGTGAGG-3′ |
| β-Actin | RT-PCR | 5′- GGCTACAGCTTCACCACCAC-3′ | 5′- TACTTGCGCTCAGGAGGAGC-3′ |
| ABCG2 | Q-PCR | 5′- GAGCCTACAACTGGCTTAGACTCAA-3′ | 5′- TGATTGTTCGTCCCTGCTTAGAC-3′ |
| β-Actin | Q-PCR | 5′-TGTCCACCTTCCAGCAGATGT-3′ | 5′-AGTCCGCCTAGAAGCATTTGC-3′ |
| GAPDH | Q-PCR | 5′- CCACCCATGGCAAATTCC-3′ | 5′- TGGGATTTCCATTGATGACAAG-3′ |
PCR to determine APOE alleles.
Genomic DNA samples were isolated from cerebellum tissues of the patients following a standard procedure as described (Hixson and Vernier, 1990). Briefly, the tissues were digested with proteinase K and DNA samples were extracted with phenol-chloroform and precipitated with ethanol. PCR was performed on DNA samples and HhaI restriction digestion was used to distinguish different apoE genotypes as described (Hixson and Vernier, 1990; Xiong et al., 2008).
Western blot analysis.
Protein samples were prepared from human and mouse brain tissues and cultured cells using extraction buffer containing 50 mm Tris-HCl, pH 7.4, 0.5% SDS and protease inhibitor cocktail P8340 (Sigma), and Western blots were performed as described previously (Zhang et al., 2003, 2006). The proteins (40 μg) from each sample were separated on a 10% SDS polyacrylamide gel (SDS-PAGE) and transferred to a piece of nitrocellulose membrane. ABCG2 protein was detected by a 1:200 dilution of a primary mouse monoclonal anti-human ABCG2 antibody (s.c.-18841, Santa Cruz Biotechnology) or a 1:1000 dilution of a primary mouse monoclonal anti-human BCRP (BXP-21) antibody followed by a 1:3000 dilution of a secondary donkey anti-mouse IgG-HRP antibody (s.c.-2314, Santa Cruz Biotechnology). β-actin protein was detected by a 1:2500 dilution of a primary mouse anti-human β-actin antibody (A5441, Sigma) followed by a 1:3000 dilution of a secondary donkey anti-mouse IgG-HRP antibody (s.c.-2314, Santa Cruz Biotechnology). ECL Plus reagents (Amersham Biosciences) were applied to the blots and the blots were exposed to autoradiography films (Kodak) for 30 s to 3 min.
Coimmunoprecipitation.
Human brain lysates were prepared as described previously (Xiong et al., 2008). Protein concentration was determined using a DC Bio-Rad protein assay kit following the manufacturer's instructions. Protein lysate (1 mg) was placed in a 1.5 ml-tube and diluted in 1× PBS buffer to 0.80 mg/ml final concentration. Synthetic Aβ1–40 peptide (5 μg) was added to each sample followed by rotation overnight at 4°C. Two tubes were prepared, and one of them was used as a negative control. Ten micrograms of a mouse monoclonal anti-Aβ IgG antibody (6E10, Signet) was added to one of the two tubes and not to the negative control. The mixtures were rotated 2–3 h at 4°C. Then, 50 μl of Protein A/G beads (Santa Cruz Biotechnology) was added, and the samples were rotated for another 2 h at 4°C. Tubes were centrifuged at 17,900 × g for 30 s and supernatant was removed. Beads were washed five times with 1 ml of 1× PBS and spun down each time for 30 s at 4°C. At last, the buffer was removed and the pellet was resuspended in 50 μl of 2× Western loading buffer. The eluted samples were resolved on a 10% SDS-PAGE and the separated proteins were transferred to nitrocellulose membrane. The blots were probed with an ABCG2 antibody BXP-21 (1:1000 dilution) followed by a 1:3000 dilution of a secondary goat anti-mouse IgG-HRP antibody (A4416, Sigma). ECL Plus reagents (Amersham Biosciences) were applied to the blots, and the blots were exposed to autoradiography films (Kodak) for 30 s to 3 min.
Cloning and expression of human ABCG2.
A full-length cDNA encoding human ABCG2 was cloned into pcDNA3.1 vector as we described previously (Zhang et al., 2003). The full-length cDNA was cleaved from pcDNA3.1 vector by digestion with EcoRI and BamHI and then subcloned into an expression vector pTT5SH8Q2 cleaved with the same restriction enzymes (the vector was generally provided by Dr. Y. Durocher at the NRC–Biotechnology Research Institute, Montreal, Quebec, Canada). The construct was sequenced for sequence accuracy and in-frame. DNA plasmids (endotoxin-free) were purified by using a QIAGEN kit following the manufacturer's instructions. The construct was transfected into HEK293 cells by using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions.
Drug uptake assay.
HEK293 cells were grown to 70% confluence in 24-well plates and transfected with hABCG2 construct or an empty vector by using Lipofectamine 2000 following the manufacturer's instructions. The cells were recovered for 48 h after transfection and used for drug uptake assays. Overexpression of ABCG2 in cells was confirmed by immunocytochemistry and Western blotting. Drug uptake assays for Hoechst 33342 were performed in the presence or absence of 5 μm synthetic Aβ1–40 peptides. The cells were incubated with or without Aβ1–40 peptides for 1 h, and then incubated with an ABCG2 substrate Hoechst 33342 (10 μg/ml) for 1 h. The cells were then washed three times with phenol red-free HBSS. Phenol red-free HBSS was added to the cells for fluorescence reading. The amount of the drug accumulated in the cells was determined by measuring fluorescence with a microplate reader (346 nm for excitation and 460 nm for emission) (FLx800, BioTek Instruments) and normalized to fluorescent units/μg protein. To determine whether Cy5.5 is a substrate of ABCG2, an equal amount of Cy5.5-free dye was added to control and ABCG2-overexpressing cells. Uptake of Cy5.5-free dye in the cells was measured by the microplate reader (673 nm for excitation and 692 nm for emission). Synthetic Aβ1–40 peptides were labeled with Cy5.5 using a kit (Cy5.5 Mono NHS ester) purchased from GE Healthcare Biosciences/Amersham following the manufacturer's instructions as described below. The labeled peptides were applied to control and ABCG2-overexpressing cells for 1 h and the cells were then washed three times with phenol red-free HBSS. Uptake of Aβ1–40 peptides was determined by measuring Cy5.5 fluorescence in cells. For each experiment, the treatment was duplicated or triplicated. The experiment was repeated for at least three times.
Optical imaging analysis of Aβ1–40 transport at the BBB in Abcg2 knock-out and wild-type mice.
Synthetic Aβ1–40 or scrambled Aβ40–1 peptides (0.5 mg) (molecular weight 4329.86) were labeled with Cy5.5 (molecular weight 1128.4) using a kit (Cy5.5 Mono NHS ester) purchased from GE Healthcare Biosciences/Amersham following the manufacturer's instructions. The labeled peptides were purified using a column Microcon Ultracel YM-3 (Regenerated cellulose 3000 MWCO) (Millipore) to remove unincorporated free dye. The amount of labeled peptides was quantified by using a BCA Protein Assay kit (Thermo Scientific) following the manufacturer's instructions and the labeling efficiency was determined by the BioTek FLx800 microplate reader (673 nm for excitation and 692 nm for emission). Adult Abcg2 KO and wild-type mice of the same age, sex, and body weight were paired. One week before the experiment, the paired mice were placed into cages with bedding that does not generate in vivo fluorescence if ingested by the mice. The fur of the mice was shaved just before the experiments under inhaled isoflurane anesthesia. Equal amounts of Cy5.5-labeled peptides (65 or 200 μg) or equal fluorescent intensity of Cy5.5-free dye were intravenously injected via tail vein into the paired mice according to experimental design. The mice were scanned alive at 15 min, 2, 4, 6, 8, and/or 24 h after injection with an optical imaging system eXplore Optix (GE Healthcare Systems/ART) as described previously (Abulrob et al., 2007, 2008). The resolution was set at 1.5 mm for mouse head. Four pairs of mice were killed at 8 and 24 h after injection, respectively. The mice were perfused with saline (40–50 ml) and brains and other organs were collected. The ex vivo brains were scanned with the optical imager at a resolution of 1 mm. In all imaging experiments, a 670 nm pulsed laser diode with a repetition frequency of 80 MHz and a time resolution of 12 ps was used for excitation. The fluorescence emission at 700 nm was collected by a highly sensitive photomultiplier tube offset by 3 mm for diffuse optical topography reconstruction (Abulrob et al., 2007, 2008). Each animal was positioned prone on a plate that was then placed on a heated plate (at 36°C) in the imaging system. Cy5.5 fluorescence in the heads and ex vivo brains was analyzed for its concentration and intensity using the software ART Optix Optiview (ART) (Abulrob et al., 2007, 2008). The brain samples were then frozen-sectioned and immunostained with the anti-Aβ antibody 6E10 to detect the presence of Aβ peptides.
Statistical analysis.
Each of the in vitro experiments or assays was duplicated or triplicated. The experiments or assays were repeated at least three times. Results are expressed as mean ± SD. The comparison between two samples was analyzed by Student's t test and multiple samples were compared by one-way ANOVA (and nonparametric). p < 0.05 is considered as significance.
Results
Patients' information
The average age of 12 ND, 13 AD, and 13 AD/CAA patients was 88.08 ± 5.24, 82.85 ± 10.09, and 83.85 ± 7.02 years, respectively. The brain samples were harvested between 1 and 3.3 h after patients' deaths, freshly frozen in liquid nitrogen and then stored at −80°C until use. Since several studies suggested that ApoE4 may play a role in Aβ deposition in cerebral microvessels (Chalmers et al., 2003; Fryer et al., 2005; Oddo et al., 2009), we analyzed apoE genotypes of the Alzheimer's patients and ND controls. None of the 12 ND controls carried an apoE4 allele. Among 13 AD/CAA patients, six carried one apoE4 allele and one carried two apoE4 allele [i.e., 54% of AD/CAA patients (7/13)]. Among 13 AD patients, five carried one apoE4 allele [e.g., 38% of AD patients (5/13)].
ABCG2 is upregulated in the brains of AD/CAA patients and in transgenic AD mice
RNA samples were isolated from AD/CAA and ND brain tissues as described above for microarray and qRT-PCR analyses. Differential expression of 273 BBB-related genes was examined by microarray analyses (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The linear regression of the microarray was R2 = 0.9292 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Four genes were differentially expressed in AD/CAA samples compared with ND brain samples (>1.33 linear fold, t test, p < 0.05) (Table. 2). ABCG2, a drug efflux transporter expressed at the BBB, was upregulated 1.58 linear fold in AD/CAA compared with ND brains (t test, p < 0.05) (Table 2). Three genes were downregulated in AD/CAA compared with ND samples, including SLC1A2 (glial high-affinity glutamate transporter) (1.42 linear fold), SLC6A6 (taurine/neurotransmitter transporter) (1.43 linear fold), and vimentin (1.51 linear fold) (Table 2). No significant changes in mRNA expression of LRP-1 and RAGE were detected in this study. The protein levels of LRP-1 and RAGE in ND and AD/CAA brains were not analyzed.
Table 2.
Microarray analyses of 273 BBB gene expression in AD/CAA versus ND brains
| Changes | Gene accession # | Gene name/function | Linear fold |
|---|---|---|---|
| Upregulation | AF098951_1 | ABCG2, BCRP | 1.57587 |
| Downregulation | NM_004171_1 | SLC1A2, glial high affinity glutamate transporter | 1.418848 |
| Downregulation | NM_003043_1 | SLC6A6, neurotransmitter transporter, taurine | 1.428843 |
| Downregulation | AK056766_1 | Vimentin | 1.510261 |
To validate ABCG2 upregulation in AD/CAA brains, real-time qRT-PCR was performed on each of the brain samples used in the microarray study using GAPDH as an internal reference control as described (Zhang et al., 2003). The analysis confirmed that the level of ABCG2 expression was over two-fold higher in AD/CAA compared with ND brains (t test, p < 0.05) (Fig. 1A). Increased expression of ABCG2 mRNA was also found in some AD brain samples, but was not statistically significant because of high variation of expression levels among different samples (Fig. 1A). Western blot analyses detected increased levels of ABCG2 protein in both AD/CAA and AD brains compared with ND brain tissues (Fig. 1B). Each lane in Figure 1B represents a mixture of six different brain samples. Microvessels were isolated from ND, AD/CAA, and AD brain tissues as described previously (Stanimirovic et al., 1997; Walker et al., 2008), and proteins were extracted from these isolated microvessels for Western blot. Increased levels of ABCG2 protein were detected in the microvessels of AD/CAA and AD brain tissues compared with ND tissues (Fig. 1C). Densitometry quantification showed that the level of ABCG2 in AD/CAA microvessels was significantly higher than that in ND (t test, p = 0.0257).
Figure 1.
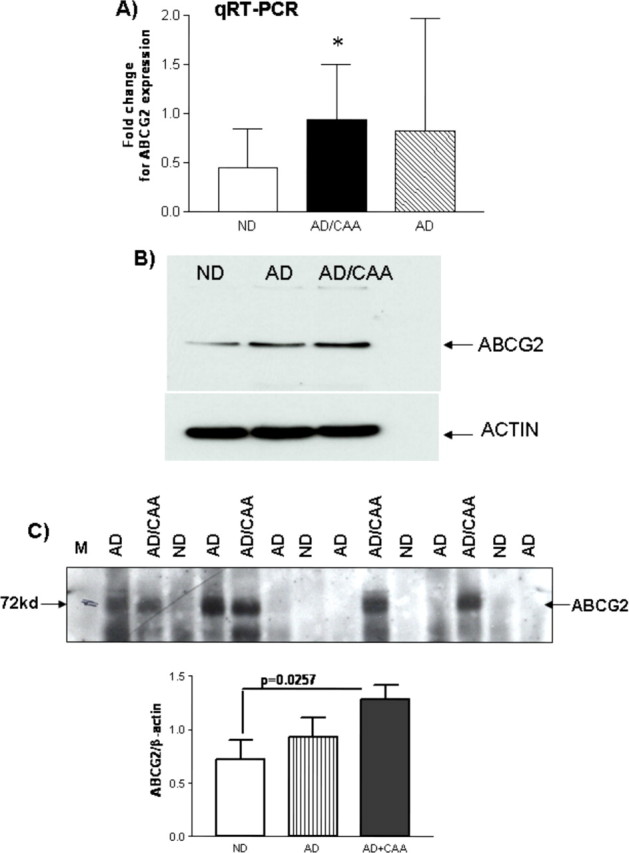
ABCG2 upregulation in human AD/CAA brains. A, Real-time quantitative RT-PCR shows that the expression of ABCG2 was significantly increased over two folds in AD/CAA brain tissues compared with that in age-matched nondemented control brains (t test, *p < 0.05). GAPDH was used as an internal reference. ABCG2 expression was also increased in AD brain samples but not statistically significant. B, Western blot shows that ABCG2 protein was upregulated in AD/CAA and AD brains compared with ND brain samples. Whole-brain homogenates were used and each lane represented the mixture of six brain samples. C, Western blot analysis of cerebral vascular protein isolated from AD, AD/CAA, and ND brains shows that ABCG2 was upregulated in cerebral vessels from AD/CAA brains (ND vs AD/CAA, t test, p = 0.0257).
Human brain tissues were frozen-sectioned for immunohistochemistry. Aβ deposits and ABCG2 expression in brain parenchyma and vessels were analyzed by staining with the anti-Aβ antibody 6E10, anti-ABCG2 antibody BXP-21, and UEA-I, respectively (Zhang et al., 2003; Mojsilovic-Petrovic et al., 2004; Xiong et al., 2008). There were no Aβ deposits and the level of ABCG2 was low or absent in ND brains (Fig. 2A,D). Aβ deposits were found in AD and AD/CAA brains and some of the deposits were associated with brain microvessels (Fig. 2B,C). The level of ABCG2 was increased in AD brain (Fig. 2E) compared with ND brain (Fig. 2D), and was also higher in AD/CAA (Fig. 2F) compared with both ND (Fig. 2D) and AD (Fig. 2E) brains. Increased ABCG2 was mainly associated with microvessels in AD and AD/CAA brains (Fig. 2E,F). AD patients had less or no Aβ deposition in cerebral microvessels and showed low levels of ABCG2 (data not shown). Overall, microarray analyses, real-time qRT-PCR, Western blots, and immunohistochemistry demonstrated that ABCG2 was upregulated in AD brains with CAA pathology.
Figure 2.

Immunohistochemical analyses of Aβ, ABCG2, and cerebral vessels in ND, AD, and AD/CAA brains. All brain sections were stained with UEA-I, and cerebral microvessels are visualized as green color. Aβ deposits and ABCG2 were detected with 6E10 and BXP-21, respectively, and visualized as red color as described in Materials and Methods. A, There was no Aβ deposition in ND brain. B, C, Aβ deposits were found in the parenchyma of both AD and AD/CAA brains, and some of the deposits were associated with microvessels. D, The level of ABCG2 was low or absent in ND brain. E, The level of ABCG2 was increased in AD compared with ND (D), and ABCG2 was mainly associated with vascular structure (green-yellowish color). F, High level of ABCG2 was found in AD/CAA microvessels compared with ND and AD brains and visualized as strong green-yellowish color.
To investigate whether Abcg2 is also upregulated in transgenic Alzheimer's mouse models, the brain samples of 3XTg and Tg-SwDI transgenic AD and relevant wild-type mice were used for immunohistochemistry and Western blot analysis. Seven pairs of Tg-SwDI and wt mouse brains (10 months of age) and six 3XTg and five wt mouse brains (at ages of 3, 6, 10, 12 and 15 months) were used in these analyses. Immunostaining with the 6E10 antibody showed high levels of Aβ peptides deposited in transgenic 3XTg AD mouse brains (Fig. 3B) but not in wt mouse brains (Fig. 3A). Aβ deposits were found in both brain parenchyma and cerebral vessels (Fig. 3D). Abcg2 was also upregulated in transgenic AD mouse brains (Fig. 3F) compared with wt controls (Fig. 3E) and was mainly associated with cerebral vessels (Fig. 3H). Western blot analyses (data not shown) demonstrated that Abcg2 was upregulated in both 3XTg and Tg-SwDI mouse brain homogenates compared with respective wt controls.
Figure 3.

Abcg2 was upregulated in transgenic AD mouse brains. Immunohistochemistry was performed on the mouse brain sections to detect the levels of Aβ deposition and Abcg2 expression. A, C, E, G, Brain sections from wild-type mice. B, D, F, H, Brain sections from the 3XTg Alzheimer's mouse brains. A and B are brain sections incubated with anti-Aβ 6E10 antibody. The 3XTg mouse brain (B) showed deposition of Aβ peptides (red color). C, D, Staining with tomato lectin (green color) overlaid with Aβ staining. Tomato lectin lit up vascular structures. Aβ deposition in the 3XTg brain (D) was found in brain parenchyma and associated with cerebral vessels. E, F, Staining with an ABCG2 antibody (red color). More ABCG2 protein was found in the 3XTg mouse brain. G, H, Vascular staining (green color) with tomato lectin overlaid with Abcg2 staining. Abcg2 in the 3XTg brain was mainly associated with cerebral vessels.
Upregulation of ABCG2 expression in human brain endothelial cells by conditioned media from Aβ-activated microglia
To examine the mechanisms by which ABCG2 is induced in the vessels of human and mouse AD brains, we investigated whether hypoxia and reoxygenation, Aβ peptides or microglial conditioned media could stimulate ABCG2 expression in HBEC cells. The cells were subjected to 1, 4 or 8 h hypoxia at 37°C in a hypoxic chamber (<2% oxygen, Anaerobic System Model 1024, Forma Scientific) equipped with a humidified, temperature-controlled incubator and subsequent reoxygenation as described previously (Zhang et al., 2006). In this and subsequent cell experiments, cells were exposed to 2–5 μm aggregated Aβ1–40 or Aβ1–42 peptides for up to 24 h (Vukic et al., 2009). The conformational state of Aβ peptides analyzed in cell media at the end of these experiments by Western blot indicated that the majority of peptides were monomers and oligomers, with a small proportion of high molecular weight aggregates (data not shown). RNA and protein samples were collected and the levels of ABCG2, HIF-1α and an internal control β-actin were analyzed by RT-PCR and Western blots. Hypoxia increased the level of HIF-1α protein, but did not affect ABCG2 expression (data not shown). The cells were then incubated with 2–5 μm Aβ1–40 or Aβ1–42 peptides or Aβ peptides in combination with hypoxia and reoxygenation. No changes in ABCG2 expression were observed in the cells under these conditions (data not shown). In our previous report (Zhang et al. 2003), astrocyte-conditioned media was shown to stimulate ABCG2 expression in HBEC. Since microglia are activated in AD brain and involved in Aβ-induced neuroinflammation (Lue et al., 2001a,b; Walker et al., 2001, 2005, 2006), we investigated whether the conditioned media from primary human microglial cultures can induce ABCG2 expression in HBEC cells. The microglial cultures were freshly isolated from postmortem human brain tissues following an established protocol (Lue and Walker, 2002) and were treated with 2 or 5 μm aggregated Aβ1–42 peptides for 24 h (Walker et al., 2001). The conditioned media were applied to HBEC cultures at 1:1 v/v ratio with HBEC medium. Real-time qPCR showed that ABCG2 expression in HBEC was strongly induced by conditioned media from Aβ-treated microglia (CM) compared with conditioned media from unstimulated microglia (one-way ANOVA, p = 0.0013) (Fig. 4). Western blotting confirmed the finding (data not shown). This indicated that paracrine factor(s) secreted from Aβ-activated microglia induced ABCG2 expression in HBEC. Previous studies described secretion of proinflammatory cytokines, such as IL-1β and TNF-α, from Aβ-treated microglia (Walker et al., 2001; Lue and Walker 2002; Hickman et al., 2008). Inflammatory cytokines have been shown to affect the expression of the BBB transporter, ABCB1/P-glycoprotein, in brain endothelial cells (Bauer et al., 2007). Therefore, we tested whether IL-1β or TNF-α could stimulate ABCG2 expression in HBEC. In the presence of 50 units of either cytokine, the expression of several inflammatory genes (including MCP-1 and IL-8) was strongly upregulated in HBEC cells (data not shown), but no changes in ABCG2 expression were observed (data not shown), suggesting that these cytokines were not involved in the microglia-mediated induction of ABCG2 in HBEC.
Figure 4.

Induction of ABCG2 expression in HBEC by conditioned media from Aβ-treated human microglia. The controls were HBEC incubated with conditioned media from unstimulated microglia. The conditioned media from primary human microglia treated with 2 μm Aβ1–42 peptides for 24 h (CM, 2 μm Aβ) or treated with 5 μm Aβ1–42 peptides for 24 h (CM, 5 μm Aβ) were applied to HBEC for 6 h. RNA samples were isolated from the cells for real-time qRT-PCR. The level of ABCG2 expression was relative to an internal control β-actin (one-way ANOVA, **p = 0.0013).
Aβ peptides interact with ABCG2 and affect ABCG2-mediated drug efflux in cells
Aβ peptides interact with membrane proteins and receptors, such as RAGE and LRP (Deane et al., 2003, 2004a, 2008; Zlokovic, 2008). To test whether Aβ peptides interact with ABCG2, synthetic Aβ1–40 peptide (5 μm) was mixed with human brain lysates and immuno-precipitated with an anti-Aβ antibody (6E10 antibody). The complexes were analyzed by Western blots using an ABCG2 antibody (BXP-21) and a secondary HRP-conjugated antibody. ABCG2 coprecipitated with Aβ1–40 peptides (Fig. 5A). Alternatively, human AD/CAA brain lysates were reacted with an ABCG2 antibody and the precipitated complexes were analyzed by Western blots using the anti-Aβ antibody 6E10, also showing that Aβ peptides coprecipitated with ABCG2 (data not shown). This demonstrated that ABCG2 in human brain lysates physically interacted with and coimmunoprecipitated with the Aβpeptides.
Figure 5.

Aβ1–40 peptides interact with ABCG2, and overexpression of cloned ABCG2 in cells. A, ABCG2 coimmunoprecipitated with Aβ1–40 peptides. Synthetic Aβ1–40 peptides (5 μm) were mixed with 1 mg of human brain lysates and then incubated with anti-Aβ 6E10 antibody. The precipitated complex were resolved on a 10% SDS-PAGE and probed with an ABCG2 antibody. Lane 1 is human brain lysate incubated with Aβ1–40 peptide and 6E10 antibody. Lane 2 is human brain lysate with Aβ1–40 peptide but not with the antibody 6E10. Lane 3 is human brain lysate after immunoprecipitated (IP) supernatant of lane 1. The co-IP demonstrates that ABCG2 in human brain interacted and coimmunoprecipitated with Aβ1–40 peptides. B, Vector map for pTT5SH8Q2 and cloning site for human ABCG2 cDNA. C, 1–4 were immunocytochemistry showing that ABCG2 protein was localized to the cell membrane of transfected cells (20×, 40×). 5 and 6 show that ABCG2 was not expressed in EGFP vector transfected cells. D, Western blot for ABCG2 protein shows that both the monomer at 72 kDa and a high-weight posttranslation-modified forms of ABCG2 were expressed in transfected HEK293 cells at 24 h after transfection (lanes 2 and 4) and highly expressed at 48 h (lanes 6 and 8). ABCG2 was not expressed in the empty vector-transfected controls (lanes 1, 3, 5, and 7) at 24 and 48 h after transfection. Lane 1, Empty vector 24 h, clone 1. Lane 2, ABCG2 vector 24 h, clone 1. Lane 3, Empty vector 24 h, clone 2. Lane 4, ABCG2 vector 24 h, clone 2. Lane 5, Empty vector 48 h, clone 1. Lane 6, ABCG2 vector 48 h, clone 1. Lane 7, Empty vector 48 h, clone 2. Lane 8, ABCG2 vector 48 h, clone 2.
To test whether Aβ peptide interaction with ABCG2 affects the drug transport function of ABCG2 in cells, hABCG2 was cloned into an expression vector (Fig. 5B). Overexpression of ABCG2 protein was confirmed in transfected cells at 24 and 48 h after transfection, and the protein was localized to the cell membrane (Fig. 5C,D). Immunocytochemistry with ABCG2 antibody (Fig. 5C) and the transfection with an enhanced green fluorescent protein (EGFP) vector (data not shown) showed that the transfection efficiency was >60%. Since ABCG2 protein was highly expressed at 48 h after transfection, this time-point was used for drug uptake assays. The cells transfected with hABCG2 or an empty vector were treated with 5 μm synthetic Aβ1–40 peptides for 1 h and then incubated with a substrate of ABCG2 Hoechst 33342 (10 μg/ml) for 1 h. The accumulation of Hoechst 33342 was significantly higher in empty-vector transfected cells than that in ABCG2-transfected cells in the absence of Aβ peptides (normalized as fluorescent units/μg protein, one-way ANOVA, p < 0.001) (Fig. 6), indicating that ABCG2 extruded the drug out of the transfected cells. However, in the presence of Aβ1–40 peptides, ABCG2-mediated drug efflux was inhibited leading to higher Hoechst 33342 accumulation in hABCG2-transfected cells (one-way ANOVA, p < 0.001) (Fig. 6). The presence of Aβ1–40 peptides did not significantly affect drug uptake in empty vector-transfected cells (data not shown). Furthermore, the uptake of Cy5.5-labeled Aβ1–40 peptides was significantly increased (34%) in hABCG2-transfected cells compared with the cells transfected with an empty vector (t test, p = 0.0003). The presence of 5 μm Aβ1–40 peptides in the media did not affect cell viability (data not shown). These results indicated that the interaction between ABCG2 and Aβ1–40 peptides interfered with the drug efflux function of ABCG2.
Figure 6.

Aβ1–40 peptides affect drug transport function of ABCG2. hABCG2-transfected HEK293 cells accumulated significantly less Hoechst 33342 than empty-vector transfected cells (one-way ANOVA test, ***p < 0.001). In the presence of Aβ1–40 peptides, hABCG2-transfected cells accumulated significantly more Hoechst 33342 compared with the controls (one-way ANOVA test, ***p < 0.001).
Abcg2 acts as a gatekeeper at the BBB for Aβ1–40 peptides
ABCG2 is a drug efflux transporter in cancer cells and at the BBB and confers multidrug resistant phenotype (Doyle and Ross, 2003; Zhang et al., 2003). However, it is not known whether ABCG2 is capable of transporting peptides. Since Aβ peptides can be transported across the BBB and ABCG2 is upregulated in AD/CAA vessels, we hypothesized that ABCG2 is involved in the clearance or transport of Aβ peptides at the BBB. To test this, we used Abcg2 knock-out mice and time-domain optical imaging analyses. Synthetic Aβ1–40 and scrambled Aβ40–1 peptides were labeled with a fluorescent dye Cy5.5, and equal amounts of the labeled peptides were injected intravenously into adult Abcg2 KO and wt mice. Since Aβ1–40 peptide is more soluble and accounts for most of the Aβ peptides deposited in cerebral vessels of Alzheimer's brain, Aβ1–40 was the peptide of choice for this study. We first tested whether Cy5.5 is a substrate of ABCG2. Cloned hABCG2 or an empty vector was transfected/overexpressed into HEK293 cells (Fig. 5C,D), as described above. An equal amount of Cy5.5-free dye was added to ABCG2- or empty-vector transfected cells for 1 h. The uptake of Cy5.5 in both control and ABCG2-overexpressing cells was similar (data not shown), suggesting that Cy5.5 is not a substrate of ABCG2. To further validate the finding, an equal amount of Cy5.5-free dye was intravenously injected into two pairs each of Abcg2 KO and wt mice. The mice were scanned alive at 15 min and 2, 4, 6, and 8 h after injection for Cy5.5 fluorescent concentration and intensity in the heads; after killing the mice at the end of the experiment, their ex vivo brains were also scanned. The analyses showed that both the fluorescent concentration and intensity were similar in the heads and ex vivo brains of Abcg2 KO and wt mice (Fig. 7), confirming that Cy5.5 is not a substrate of ABCG2 in vivo.
Figure 7.

Cy5.5 is not a substrate of ABCG2. Equal amount of Cy5.5-free dye was intravenously injected into two pairs of adult wild-type and Abcg2 KO mice. The mice were scanned alive with the optical imager at 15 min and 2, 4, 6, and 8 h. Ex vivo brains were collected at the end of experiments and scanned. The concentration of Cy5.5 in the heads of Abcg2 KO and wt mice was similar.
Eight pairs of Abcg2 KO and wt mice were then injected intravenously with an equal amount of Cy5.5-labeled Aβ1–40 peptides (200 μg/mouse) and scanned alive at 15 min, 2, 4, 6, 8, and/or 24 h after injection. Four pairs of mice were killed at either 8 or 24 h after injection, and their ex vivo brains were collected and scanned. Fluorescent concentration was significantly higher in both the heads and ex vivo brains of Abcg2 KO compared with wt mice at all scanning time-points (one-way ANOVA, p < 0.0001) (Fig. 8). Similarly, fluorescent intensity was significantly higher in both the heads and ex vivo brains of Abcg2 KO compared with wt mice (data not shown). Two pairs of Abcg2 KO and wt mice injected with lower concentration of Cy5.5-labeled Aβ1–40 peptides (65 μg/mouse) showed essentially the same result: comparatively higher brain fluorescence concentration in Abcg2 KO than in wt animals, although at overall lower fluorescence intensity (data not shown).
Figure 8.

Abcg2 KO mice injected with Cy5.5-labeled Aβ1–40 peptides show increased fluorescence concentration in the heads and brains. Equal amounts of Cy5.5-labeled Aβ1–40 were injected intravenously into 8 pairs of adult wt and Abcg2 KO mice. A, Mice were scanned alive at 15 min, 2, 4, 6, 8, and/or 24 h after injection. Higher fluorescence was found in the heads of the Abcg2 KO mice compared with wt mice at different time points. B, The mice were killed at 8 or 24 h after injection and the ex vivo brains were scanned. Higher Cy5.5 fluorescence was found in the ex vivo brains of Abcg2 KO mice compared with wt mice. C, The average of Cy5.5 fluorescence was significantly higher in the heads of Abcg2 KO mice compared with wt mice at different time points (one-way ANOVA, ***p < 0.0001).
To investigate whether the observed effect was specific to Aβ peptides, Cy5.5-labeled scrambled Aβ40–1 peptides were intravenously injected into two pairs of Abcg2 KO and wt mice. The mice were scanned alive at 15 min and 2, 4, 6, and 8 h after injection. The analyses showed no statistical difference between Abcg2 KO and wt animals (data not shown). This demonstrated that the observed effect of Abcg2 on Aβ transport at the BBB was specific to Aβ1–40 peptides but not to scrambled Aβ40–1 peptides.
To further validate this finding, two pairs of Abcg2 KO mice were injected intravenously with either Cy5.5-free dye or Cy5.5-labeled Aβ1–40 peptides at equal fluorescent intensity. Cy5.5 fluorescent concentration (as well as fluorescence intensity) were significantly higher in the heads and the ex vivo brains of Abcg2 KO mice injected with Cy5.5-labeled Aβ1–40 peptides (159.38%) compared with Abcg2 KO mice injected with Cy5.5-free dye (100%) (t test, p < 0.001) (Fig. 9), suggesting that the BBB in Abcg2 KO mice was intact for Cy5.5-free dye molecule (1 kDa), and that Cy5.5-labeled Aβ1–40 peptides were actively transported into the brain in the absence of Abcg2 at the BBB.
Figure 9.
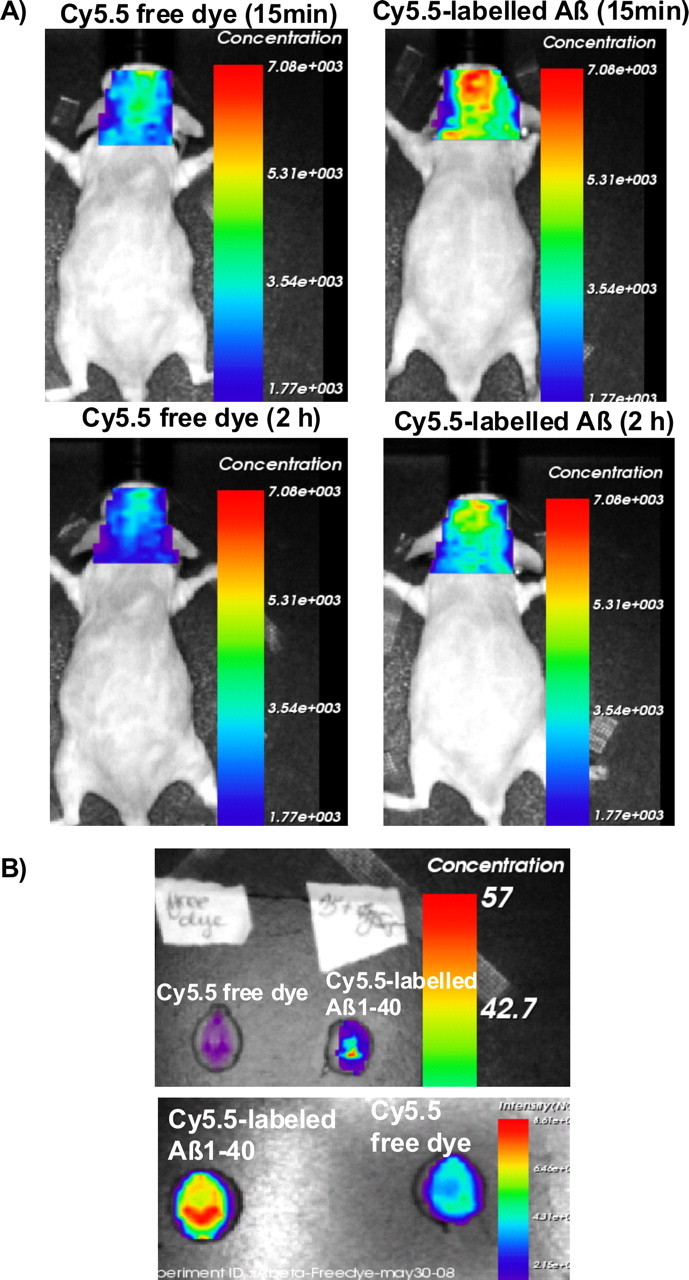
Absence of Abcg2 allows more Aβ peptides to be transported into the brain. A, Two pairs of Abcg2 KO mice were injected intravenously Cy5.5-free dye or Cy5.5-labeled Aβ1–40 peptides in equal fluorescence intensity. The animals were scanned alive for 15 min and 2, 4, 6, and 8 h. B, Ex vivo brains collected at the end of experiment were scanned. Cy5.5 fluorescence concentration was significantly higher in the brains of Abcg2 KO mice injected with Cy5.5-labeled Aβ peptides (159.38%) compared with these injected with Cy5.5-free dye (100%) (t test, p < 0.001). This demonstrates that the Cy5.5 was brought into the brain as a form of Cy5.5-labeled Aβ1–40 peptides, indicating that Abcg2 is required at the BBB to prevent circulating Aβ peptides from entry into the brain.
To further show that Cy5.5-Aβ peptides accumulated in Abcg2 KO mouse brains after intravenous injection, the brain tissues were immunostained with the anti-Aβ antibody 6E10 (Fig. 10). Aβ immunofluorescence was strongly enhanced in the brains of Abcg2 KO mice compared with wt mice (Fig. 10C,D).
Figure 10.

Abcg2 KO mice accumulate more Aβ peptides in the brains. A, B, Brain sections from wild-type mice. C, D, Brain sections from Abcg2 KO mice. Both mice were injected with Cy5.5-labeled Aβ1–40 peptides. A, C, The brain sections were stained only with secondary antibody but not the Aβ antibody. B, D, The brain sections were stained with Aβ antibody (6E10) and the secondary antibody. More Aβ fluorescence was found in Abcg2 KO mouse brain (D).
Discussion
The accumulation and deposition of Aβ peptides in the brain and around cerebral vessels is one of the main pathological characteristics of Alzheimer's disease. A number of in vitro and in vivo studies have shown that Aβ peptides are involved in Alzheimer's pathogenesis and synaptic dysfunction. It has been proposed that Aβ accumulation in the brain and cerebral vessels results from both brain over-production and aberrant clearance/transport of these peptides across the blood brain barrier (BBB) (Zlokovic, 2008). The BBB is both physical and biological barrier that restricts the passage of blood-borne substances (such as drugs and neurotoxins) while facilitating the transport of nutrients (such as amino acids, glucose, and nucleosides) into the brain, and is therefore critical for maintaining CNS homeostasis (Zhang and Stanimirovic, 2005). The integrity of the BBB is compromised in Tg2576 Alzheimer's mice as early as 4 months of age (Dickstein et al., 2006), and the active or passive immunization against Aβ peptides restores the BBB permeability in this model of AD (Dickstein et al., 2006). It was suggested that Aβ peptides can be transported across the BBB and that disrupted vascular clearance of Aβ peptides from the brain may contribute to their accumulation in AD (Zlokovic, 2008). Recent studies provided the evidence that several proteins and receptors, including LRP-1, sLRP, ApoE2, ApoE3, and ApoJ, may be involved in clearing Aβ peptides from the brain (Deane et al., 2004a, 2008a,b; Bell et al., 2007; Sagare et al., 2007). Endothelial RAGE, instead, has been shown to transport Aβ peptides from the circulation into the brain (Deane et al., 2004b). The increased density of both LRP-1 and RAGE-positive brain capillaries has been observed in AD (Jeynes and Provias, 2008). The levels of LRP-1 in capillaries are inversely related to the degree of Aβ burden in the AD brains.
To examine the expression of BBB-related genes in AD/CAA brains, we developed a custom BBB microarray which represents 273 human genes related to transport and metabolic functions of the BBB. Differential expressional profiling between AD/CAA and ND brains using this microarray identified a significant upregulation of the BBB efflux transporter, ABCG2, in AD/CAA brain. The upregulation of ABCG2 in the brain and brain vessels of AD/CAA patients was subsequently confirmed by real-time qRT-PCR, Western blots, and immunohistochemistry. Increased Abcg2 expression was also observed in the brains and/or cerebral vessels of transgenic 3XTg and Tg-SwDI mouse AD models. Tg-SwDI mouse model carries a human APP gene with mutations that specifically generate a vasculotropic Dutch/Iowa mutant form of β-amyloid protein precursor (Davis et al., 2004). These transgenic mice rapidly develop early-onset and cerebral microvascular accumulation of β-amyloid peptides (Davis et al., 2004) and serve as a good model to study cerebral amyloid angiopathy (Davis et al., 2004, 2006; Miao et al., 2005; Van Vickle et al., 2008). The 3XTg mouse model of Alzheimer's disease recapitulates both Aβ and tau pathologies (Oddo et al., 2003) observed in human AD (Oddo et al., 2003). Increased Abcg2 expression was found in the brains and cerebral vessels of the 3XTg AD mice at ages of 3, 6, 10, 12 and 15 months. Collectively, these findings suggested that increased ABCG2 expression may be involved in Aβ trafficking at the BBB.
Although ABCG2 expression in AD/CAA appears to correlate with the accumulation and deposition of Aβ in cerebral vessels, potential mechanisms involved in the regulation of this gene in cerebral vessels remain unclear. Brain vessels affected with CAA are dysfunctional and suffer from hypoxic conditions and inflammation that jointly promote angiogenic transformation (Vagnucci and Li, 2003). The experimental evidence presented in this study suggested that neither Aβ peptides alone nor in combination with hypoxic microenvironment stimulated ABCG2 expression in human brain endothelial cells. In contrast, conditioned media from Aβ-activated microglia stimulated ABCG2 expression in brain endothelial cells, suggesting that paracrine factors released from activated microglia may be responsible for increased ABCG2 expression in AD brain. Microglia are activated in Alzheimer's brain and under cultured conditions by Aβ peptides (Lue et al., 2001a,b, 2002; Walker et al., 2001, 2005, 2006). In addition, RAGE has been shown to control trafficking of mononuclear macrophages and monocytes across the BBB, which might increase the pool of perivascular microglia (Giri et al., 2000), thus amplifying influence on BBB expression of Aβ receptors and inflammation-driven angiogenesis. The nature of secreted microglia factors responsible for inducing ABCG2 expression in HBEC is presently unclear, although preliminary evidence suggested that cytokines IL-1β and TNFα are not important effectors. Recent studies reporting that IL-6 or endoplasmic reticulum (ER) stress induce ABCG2 expression in plasma cells (Nakamichi et al., 2009) and that IL-6 is upregulated in HBEC treated with Aβ1–40 peptides, as well as in AD and AD/CAA brains (Vukic et al., 2009), suggest IL-6 as a potential mediator of ABCG2 upregulation in AD. Since it is difficult to recreate in vivo chronic exposure of the multicellular neurovascular unit to Aβ peptides, hypoxia, and inflammatory microenvironment in cell culture models, we cannot rule out the possibility that the combination of these factors might be responsible for stimulation of vascular ABCG2 expression in AD. Furthermore, given that AD and brain vascular diseases (i.e., stroke, small vessel disease) have a significant comorbidity and share the same risk factors (i.e., hypertension, diabetes, hypercholesterolemia) (Jellinger, 2002), we cannot completely exclude the possibility that the observed upregulation of ABCG2 in brain vessels in AD/CAA might be associated with vascular pathology not directly related to AD.
The evidence of physical interaction between Aβ peptides and ABCG2 protein shown in our experiments suggested that ABCG2 may be involved in the process of Aβ trafficking across the BBB. Furthermore, in vivo experiments demonstrated that the absence of Abcg2 allowed ∼20–37% more Aβ1–40, but not the scrambled Aβ40–1 peptides, to be transported into the brain. This suggested that ABCG2 may act as a gatekeeper at the BBB that prevents circulatory Aβ peptides from entering into the brain, as proposed in the model shown in Figure 11. The model suggests that, in addition to transporters involved in luminal-to-abluminal and abluminal-to-luminal transport of Aβ across the BBB, specific luminal BBB efflux pumps could also participate in maintaining Aβ homeostasis by preventing access of circulatory Aβ into the brain. Interestingly, ABCB1/MDR-1 P-glycoprotein (Pgp), a major drug efflux transporter expressed at the luminal interface of the BBB, has been shown to interact with Aβ peptides and to efflux Aβ peptides in vitro (Lam et al., 2001). Furthermore, studies in transgenic animals demonstrated that lack of Pgp at the BBB reduces the clearance rate of Aβ peptides injected into the brain, as well as that APP-transgenic/Pgp-null mice exhibit enhanced Aβ deposition compared with APP-transgenic/Pgp wt mice (Cirrito et al., 2005) (Fig. 11). These studies suggested that luminal BBB transporter could efflux parenchymal Aβ. Although the mechanisms of such function are not completely clear, these studies imply that ABCG2, in addition to efflux of circulatory Aβ described in this study, might also be able to efflux parenchymal Aβ (Fig. 11), but further investigation is needed to examine this possibility. In addition, abluminal LRP-1, sLRP, ApoE2/E3, and ApoJ have also been implicated in the clearance of Aβ peptides from the brain (Shibata et al., 2000; Deane et al., 2004a, 2008a,b; Bell et al., 2007; Sagare et al., 2007) (Fig. 11).
Figure 11.

The trafficking of Aβ peptides across the blood–brain barrier. The roles of various transporters and receptors in Aβ transport or clearance at the BBB are depicted according to the literature and our experimental evidence.
This study provides the evidence of the association/correlation of ABCG2 expression in brain vessels with CAA in human AD, as well as with Aβ deposition in transgenic animal models. Since demonstrated functional role of ABCG2 at the BBB is efflux of circulatory Aβ, the link between ABCG2 upregulation and vascular accumulation of Aβ remains speculative. ABCG2 upregulation could be a compensatory mechanism initiated by pathological microenvironment in the neurovascular unit, aimed at reducing Aβ burden in the brain by preventing access of circulatory Aβ into the brain. In contrast, it could be an early vascular change involved in maintaining circulatory pool of Aβ and thus preventing Aβ clearance from perivascular or parenchymal pools. Passive and active immunization strategies for AD proposed that depleting circulatory pool of Aβ could mobilize Aβ clearance from the brain (DeMattos et al., 2001, 2002; Solomon, 2007). Although this hypothesis assumes equilibrium of circulatory and brain Aβ pools at the macro level, it is plausible that this equilibrium could be controlled in “micro” domains of single vessel or groups of vessels where changes in the expression of Aβ transporters could create disequilibrium that facilitates or prevents Aβ clearance from the brain. Finally, as observed in the in vitro studies, binding of circulatory Aβ peptides to ABCG2 may competitively inhibit the binding of substrate drugs and contribute to altered BBB permeability in AD patients.
In summary, the study describes ABCG2 upregulation in cerebral vessels of human and mouse AD brains, suggesting that ABCG2 upregulation may be a biomarker of CAA vascular pathology in AD. In culture models, ABCG2 expression in brain endothelial cells was upregulated by paracrine factor(s) released from Aβ-activated microglia. The interaction of Aβ1–40 peptides with ABCG2 was shown to interfere with ABCG2-mediated drug transport. In Abcg2 knock-out animals, we observed increased transport and accumulation of Aβ peptides in the brain, suggesting that Abcg2 may play a role of gatekeeper at the BBB to prevent circulatory Aβ peptides from entering into the brain.
Footnotes
This study was supported by funding from the Canadian Research Program “Vascular Health and Dementia” sponsored by the Heart and Stroke Foundation of Canada, Canadian Institute of Health Research, Alzheimer Society of Canada, and Pfizer, and the funding from the National Research Council of Canada. The research work in L.-F.L.'s laboratory was supported by a National Institutes of Health RO1 grant (RO1 NS049075). The Sun Health Research Institute Brain Donation Program of Sun City, AZ, was supported by the National Institute on Aging (Grant P30 AG19610, Arizona Alzheimer's Disease Core Center), the Arizona Department of Health Services (Contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (Contracts 4001, 0011, and 05-901 to the Arizona Parkinson's Disease Consortium), and the Prescott Family Initiative of the Michael J. Fox Foundation for Parkinson's Research. We would like to thank Dr. B. Chakravarthy and T. Atkinson [National Research Council–Institute for Biological Sciences (NRC–IBS)] and Dr. C. Wellington and V. Hirsch-Reinshagen (University of British Columbia) for providing transgenic Alzheimer's mouse brain samples, Dr. A. Prat (Montreal Neurological Institute) for providing primary human brain endothelial cells, Dr. Y. Durocher (NRC–Biotechnology Research Institute) for providing the cloning vector pTT5SH8Q2, Dr. Seema Haqqani (NRC–IBS) for compiling the list of the genes on the custom BBB microarray, B. Smith and J. LeBlanc (NRC–IBS) for designing and printing the custom BBB microarray slides, Dr. A. Abulrob, E. Brunette, Dr. U. Iqbal, and Max London (NRC–IBS) for their help in the imaging work, and NRC–IBS Animal Facility staff. We are grateful to the Sun Health Research Institute Brain Donation Program for the provision of human brain tissue. J.B. is a visiting scholar from the Shanxi Medical University, China. K.P. is a visiting scholar from the hospital affiliated with the Shandong University of Traditional Chinese Medicine, China.
References
- Abulrob A, Brunette E, Slinn J, Baumann E, Stanimirovic D. In vivo time domain optical imaging of renal ischemia-reperfusion injury: discrimination based on fluorescence lifetime. Mol Imaging. 2007;6:304–314. [PubMed] [Google Scholar]
- Abulrob A, Brunette E, Slinn J, Baumann E, Stanimirovic D. Dynamic analysis of the blood–brain barrier disruption in experimental stroke using time domain in vivo fluorescence imaging. Mol Imaging. 2008;7:248–262. [PubMed] [Google Scholar]
- Bauer B, Hartz AM, Miller DS. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood–brain barrier. Mol Pharmacol. 2007;71:667–675. doi: 10.1124/mol.106.029512. [DOI] [PubMed] [Google Scholar]
- Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27:909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers K, Wilcock GK, Love S. APOE epsilon 4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of Abeta protein. Neuropathol Appl Neurobiol. 2003;29:231–238. doi: 10.1046/j.1365-2990.2003.00457.x. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM. P-glycoprotein deficiency at the blood–brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Davis J, Xu F, Miao J, Previti ML, Romanov G, Ziegler K, Van Nostrand WE. Deficient cerebral clearance of vasculotropic mutant Dutch/Iowa Double A beta in human A betaPP transgenic mice. Neurobiol Aging. 2006;27:946–954. doi: 10.1016/j.neurobiolaging.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004a;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood–brain barrier. Stroke. 2004b;35(11) Suppl 1:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Zlokovic BV. The role of the cell surface LRP and soluble LRP in blood–brain barrier Abeta clearance in Alzheimer's disease. Curr Pharm Des. 2008a;14:1601–1605. doi: 10.2174/138161208784705487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008b;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- Dickstein DL, Biron KE, Ujiie M, Pfeifer CG, Jeffries AR, Jefferies WA. Abeta peptide immunization restores blood–brain barrier integrity in Alzheimer disease. FASEB J. 2006;20:426–433. doi: 10.1096/fj.05-3956com. [DOI] [PubMed] [Google Scholar]
- Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri R, Shen Y, Stins M, Du Yan S, Schmidt AM, Stern D, Kim KS, Zlokovic B, Kalra VK. beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol. 2000;279:C1772–1781. doi: 10.1152/ajpcell.2000.279.6.C1772. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- Jeynes B, Provias J. Evidence for Altered LRP/RAGE Expression in Alzheimer Lesion Pathogenesis. Curr Alzheimer Res. 2008;5:432–437. doi: 10.2174/156720508785908937. [DOI] [PubMed] [Google Scholar]
- Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, Greenberg SM. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007;68:1411–1416. doi: 10.1212/01.wnl.0000260066.98681.2e. [DOI] [PubMed] [Google Scholar]
- Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB. Beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem. 2001;76:1121–1128. doi: 10.1046/j.1471-4159.2001.00113.x. [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker DG. Modeling Alzheimer's disease immune therapy mechanisms: interactions of human postmortem microglia with antibody-opsonized amyloid beta peptide. J Neurosci Res. 2002;70:599–610. doi: 10.1002/jnr.10422. [DOI] [PubMed] [Google Scholar]
- Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Jr, Brachova L, Yan SD, Walker DG, Shen Y, Rogers J. Inflammatory repertoire of Alzheimer's disease and nondemented elderly microglia in vitro. Glia. 2001a;35:72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer's disease with human postmortem microglial cultures. Neurobiol Aging. 2001b;22:945–956. doi: 10.1016/s0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- Miao J, Xu F, Davis J, Otte-Höller I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Nesic M, Pan A, Zhang W, Stanimirovic D. Development of rapid staining protocols for laser capture microdissection of brain vessels from human and rat coupled to gene expression analyses. J Neurosci Methods. 2004;133:39–48. doi: 10.1016/j.jneumeth.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Nakamichi N, Morii E, Ikeda JI, Qiu Y, Mamato S, Tian T, Fukuhara S, Aozasa K. Synergistic effect of interleukin-6 and endoplasmic reticulum stress inducers on the high level of ABCG2 expression in plasma cells. Lab Invest. 2009;89:327–336. doi: 10.1038/labinvest.2008.157. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Cheng D, Laferla FM. Genetically altering Abeta distribution from the brain to the vasculature ameliorates tau pathology. Brain Pathol. 2009 doi: 10.1111/j.1750-3639.2008.00194.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, Thal LJ. The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer's disease and Lewy body variant. Neurology. 1996;47:190–196. doi: 10.1212/wnl.47.1.190. [DOI] [PubMed] [Google Scholar]
- Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol. 2003;29:106–117. doi: 10.1046/j.1365-2990.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13:1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer's disease: Molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood–brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon B. Antibody-mediated immunotherapy for Alzheimer's disease. Curr Opin Investig Drugs. 2007;8:519–524. [PubMed] [Google Scholar]
- Stanimirovic D, Shapiro A, Wong J, Hutchison J, Durkin J. The induction of ICAM-1 in human cerebromicrovascular endothelial cells (HCEC) by ischemia-like conditions promotes enhanced neutrophil/HCEC adhesion. J Neuroimmunol. 1997;76(1–2):193–205. doi: 10.1016/s0165-5728(97)00057-x. [DOI] [PubMed] [Google Scholar]
- Vagnucci AH, Jr, Li WW. Alzheimer's disease and angiogenesis. Lancet. 2003;361:605–608. doi: 10.1016/S0140-6736(03)12521-4. [DOI] [PubMed] [Google Scholar]
- Van Vickle GD, Esh CL, Daugs ID, Kokjohn TA, Kalback WM, Patton RL, Luehrs DC, Walker DG, Lue LF, Beach TG, Davis J, Van Nostrand WE, Castaño EM, Roher AE. Tg-SwDI transgenic mice exhibit novel alterations in AbetaPP processing, Abeta degradation, and resilient amyloid angiopathy. Am J Pathol. 2008;173:483–493. doi: 10.2353/ajpath.2008.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukic V, Callaghan D, Walker D, Lue LF, Liu QY, Couraud PO, Romero IA, Weksler B, Stanimirovic DB, Zhang W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer's brain is mediated by the JNK-AP1 signaling pathway. Neurobiol Dis. 2009;34:95–106. doi: 10.1016/j.nbd.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Lue LF. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer's disease and other neurodegenerative diseases. J Neurosci Res. 2005;81:412–425. doi: 10.1002/jnr.20484. [DOI] [PubMed] [Google Scholar]
- Walker DG, Lue LF, Beach TG. Gene expression profiling of amyloid beta peptide-stimulated human post-mortem brain microglia. Neurobiol Aging. 2001;22:957–966. doi: 10.1016/s0197-4580(01)00306-2. [DOI] [PubMed] [Google Scholar]
- Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- Walker DG, Dalsing-Hernandez JE, Lue LF. Human postmortem brain-derived cerebrovascular smooth muscle cells express all genes of the classical complement pathway: a potential mechanism for vascular damage in cerebral amyloid angiopathy and Alzheimer's disease. Microvasc Res. 2008;75:411–419. doi: 10.1016/j.mvr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker PR, Smith B, Liu QY, Famili AF, Valdés JJ, Liu Z, Lach B. Data mining of gene expression changes in Alzheimer brain. Artif Intell Med. 2004;31:137–154. doi: 10.1016/j.artmed.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Weller RO, Nicoll JA. Cerebral amyloid angiopathy: pathogenesis and effects on aging and Alzheimer brain. Neurol Res. 2003;25:611–616. doi: 10.1179/016164103101202057. [DOI] [PubMed] [Google Scholar]
- Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, Sue LI, Woulfe J, Xu H, Stanimirovic DB, Zhang W. Cholesterol retention in Alzheimer's brain is responsible for high β-and γ-secretase activities and Aβ production. Neurobiol Dis. 2008;29:422–437. doi: 10.1016/j.nbd.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Stanimirovic DB. The transport systems of the blood–brain barrier. In: De Vries E, Prat A, editors. The blood–brain barrier and its microenvironment: basic physiology to neurological disease. New York: Taylor and Francis Group; 2005. pp. 103–142. [Google Scholar]
- Zhang W, Mojsilovic-Petrovic J, Andrade MF, Zhang H, Ball M, Stanimirovic DB. Expression and functional characterization of ABCG2 in brain endothelial cells and vessels. FASEB J. 2003;17:2085–2087. doi: 10.1096/fj.02-1131fje. [DOI] [PubMed] [Google Scholar]
- Zhang W, Petrovic JM, Callaghan D, Jones A, Cui H, Howlett C, Stanimirovic D. Evidence that hypoxia-inducible factor-1 (HIF-1) mediates transcriptional activation of interleukin-1β (IL-1β) in astrocyte cultures. J Neuroimmunol. 2006;174:63–73. doi: 10.1016/j.jneuroim.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Cerebrovascular transport of Alzheimer's amyloid beta and apolipoproteins J and E: possible anti-amyloidogenic role of the blood–brain barrier. Life Sci. 1996;59:1483–1497. doi: 10.1016/0024-3205(96)00310-4. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]