




Abstract
Hydrogen/deuterium exchange measurements by mass spectrometry (HX-MS) can be used to report localized conformational mobility within folded proteins, where exchange predominantly occurs through low energy fluctuations in structure, allowing transient solvent exposure. Changes in conformational mobility may impact protein function, even in cases where structural changes are unobservable. Previous studies of the MAP kinase, ERK2, revealed increases in HX upon activation occured at the hinge between conserved N- and C-terminal domains, which could be ascribed to enhanced backbone flexibility. This implied that kinase activation modulates interdomain closure, and was supported by evidence for two modes of nucleotide binding that were consistent with closed vs open conformations in active vs inactive forms of ERK2, respectively. Thus, phosphorylation of ERK2 releases constraints to interdomain closure, by modulating hinge flexibility. In this study, we examined ERK1, which shares 90% sequence identity with ERK2. HX-MS measurements of ERK1 showed similarities with ERK2 in overall deuteration, consistent with their similar tertiary structures. However, the patterns of HX that were altered upon activation of ERK1 differed from those in ERK2. In particular, alterations in HX at the hinge region upon activation of ERK2 did not occur in ERK1, suggesting that the two enzymes differ with respect to their regulation of hinge mobility and interdomain closure. In agreement, HX-MS measurements of nucleotide binding suggested revealed domain closure in both inactive and active forms of ERK1. We conclude that although ERK1 and ERK2 are closely related with respect to primary sequence and tertiary structure, they utilize distinct mechanisms for controlling enzyme function through interdomain interactions.
Keywords: hydrogen exchange, mass spectrometry, ERK1, MAP kinase, interdomain interactions
Introduction
Extracellular signal-regulated protein kinases, ERK1 and ERK2, are mitogen-activated protein (MAP) kinases which control diverse cellular responses, including cell proliferation, differentiation, transformation, and survival. ERK activation is frequently dysregulated in human cancers, due to oncogenic mutations in upstream pathway components, including Ras, Raf, and MAP kinase kinase (MKK), as well as receptor tyrosine kinases, EGFR, Her2/Neu, and Met [1,2], all which are attractive targets for drug intervention. Although ERK1 and ERK2 are functionally redundant in many cell types, each shows a distinct mouse knockout phenotype, where deletion of ERK2 leads to embryonic lethality while deletion of ERK1 yields viable animals with defects in thymocyte maturation and immunodeficiency [3,4]. Thus, at least some functions of ERK1 and ERK2 are non-redundant. On the other hand, relatively little is known about how regulatory mechanisms controlling ERK1 and ERK2 differ, which might impact the development of different strategies for specifically targeting each form.
Crystallographic studies of ERK1 and ERK2 show a conserved tertiary structure shared with all protein kinases, consisting of an N-terminal ATP binding and C-terminal substrate binding domains surrounding the catalytic site [5-8]. Both ERK1 and ERK2 are activated by dual phosphorylation at a conserved pThr-Glu-pTyr sequence located within the activation lip region, which is catalyzed by the upstream MAP kinase kinases, MKK1 and MKK2. Structural remodeling events which accompany ERK activation have been revealed by X-ray structures of inactive, unphosphorylated (0P) and active, diphosphorylated (2P) forms of ERK2 [6-8]. Diphosphorylation triggers new phosphate-side chain ion pair interactions which lead to dramatic conformational changes within the activation lip, reorganizing the substrate binding site to enable recognition of the proline-directed phosphorylation motif (pSer/pThr-Pro), and reorienting active site residues involved in catalysis.
Hydrogen-exchange mass spectrometry (HX-MS) studies have shown that protein kinases are regulated not only through changes in conformation, but also through changes in conformational mobility. In previous studies of ERK2, comparisons between the active and inactive states of this kinase revealed increased HX in the hinge region upon activation, consistent with enhanced flexibility and conformer interconversions, which were not observable by X-ray crystallography [9]. Electron paramagnetic resonance (EPR) analysis of backbone residues in ERK2 that were individually coupled to a MTSL nitroxide spin label probe demonstrated that changes in side chain correlation rates occurred upon phosphorylation and activation [10]. Taken together, the results suggested a model in which the changes in HX could be explained by regulated protein motions, such as underlying backbone flexibility. Further measurements of steric protection from HX by the ATP analogue, AMP-PNP, suggested a function for this control of hinge flexibility. On one hand, HX within the N-terminal domain was comparable between 0P-ERK2 and 2P-ERK2, in regions containing the Gly loop, the conserved Lys-Glu ion pair within β3 and αC, the hinge region, and the catalytic base, all of which are known to form close interactions with nucleotide [11]. On the other hand, 2P-ERK2 showed 10-fold greater protection from HX than 0P-ERK2 within the conserved DFG motif, part of the C-terminal domain which forms metal coordination interactions with Mg+2-ATP. Thus, AMP-PNP binding sterically protected both N- and C-terminal domains of 2P-ERK2 from solvent, but mainly protected only the N-terminal domain of 0P-ERK2. Because the two kinase forms shared similar binding affinities for AMP-PNP, the results suggested that distinct modes of nucleotide binding occur in solution. The protection patterns are consistent with a model in which 2P-ERK2 adopts a closed conformation, whereas 0P-ERK2 is somehow constrained to interfere with interdomain closure. Thus, HX protection by Mg+2-AMP-PNP binding provides a useful way to monitor domain closure, an event needed to form a competent catalytic site. These results, together with the observed changes in hinge flexibility upon activation, led us to propose that activation of ERK2 releases constraints to domain closure by increasing backbone flexibility at the hinge [11].
Given their sequence and structural conservation, different MAP kinases might be expected to show similar regulatory mechanisms. In agreement, p38α MAP kinase appears to undergo conformational rearrangements following phosphorylation which parallel those seen in ERK2. For example, structural comparisons between p38α MAP kinase in its inactive, unphosphorylated form and the related p38γ MAP kinase in its active, diphosphorylated form show large conformational changes within the activation lip following phosphorylation by MKK3/6 [12-14]. Thus, although the activation lip conformation of 0P-p38α MAP kinase differs from that of 0P-ERK2, the lip conformation of 2P-p38γ MAP kinase is similar to that of 2P-ERK2, revealing that diverse activation lip conformers in MAP kinases converge towards a uniform structure in the active state, which allows catalytic rate enhancement. In contrast, HX-MS studies show that p38α MAP kinase differs significantly from ERK2 with respect to the patterns of regional HX which change in response to kinase activation [15]. This suggests that the effect of kinase activation on conformational mobility diverges between related protein kinases, leading to the hypothesis that different MAP kinases may have distinct mechanisms for regulating enzyme activity through their control of internal protein motions.
In this study, we wished to understand whether differences in patterns of regulated conformational mobility upon activation might also be observed between enzymes that are even more closely related. Here, we analyzed ERK1, which shares 90% sequence identity with ERK2. We found that ERK1 showed significant differences in how the HX behavior changes in response to kinase activation within conserved regions of the core structure. Notably, ERK1 did not display altered HX within hinge residues as previously observed in ERK2. This suggests that ERK1 might lack constraints to interdomain closure which are present in the inactive form of ERK2. In order to test this hypothesis, we measured changes in protection from HX upon AMP-PNP binding in ERK1, and compared these to prior studies of ERK2 [11]. The results showed significant protection from HX within the C-terminal domain of both 0P-ERK1 and 2P-ERK1, indicating that both inactive and active forms appear capable of interdomain closure in solution. Thus, the occurrence and functional effects of regulated protein motions differ in substantive ways between ERK1 and ERK2.
Materials and Methods
Protein purification
Active, diphosphorylated (2P) ERK1 was produced from plasmid pET-His-ERK1-R4F, containing wild-type human His6-ERK1 expressed in tandem with constitutively active mutant MKK1 (R4F: ΔN3/S218E/S222D) [16]. Inactive, unphosphorylated (0P) human His6-ERK1 was produced using plasmid NpT7-ERK1 [17]. Each kinase form was expressed in E. coli strain BL21(DE3)-pLysS, and purified using Ni-NTA agarose (Qiagen) chromatography, desalted by gel filtration (PD10, GE Healthcare), and further purified by MonoQ FPLC. Proteins were dialyzed overnight into 50 mM KPO4 (pH 7.4), 100 mM KCl and 5 mM dithiothreitol, and stored in aliquots at −80 °C. The 0P-ERK1 protein was found to be mono-phosphorylated to 0.13 mol/mol, and was therefore dephosphorylated using λ phosphatase (New England Biolabs, 100 U/μg, 3 h, 30 C) before Mono Q separation. The activation lip phosphorylation stoichiometries of the final kinase preparations showed 0% phosphorylation in 0P-ERK1 at the regulatory Thr and Tyr residues in the activation lip, and 97% diphosphorylation in 2P-ERK1. Intact ESI-MS of 0P-ERK1 and 2P-ERK1 showed full length masses expected of the recombinant ERK1 sequences, in unphosphorylated and diphosphorylated forms (Suppl. Fig. 1).
HX-MS measurements
Data were collected on a QStar Pulsar QqTOF mass spectrometer (ABI) interfaced with an Agilent Cap1100 HPLC (0.5 mm i.d. × 10 cm column, packed with POROS R1 20 resin), and measurements of weighted average mass were performed as described [18,19]. Proteins (4 μg) were incubated in 90% D2O (Sigma) at 10 °C, allowing in-exchange reactions to take place between 8 s – 4 h. Reactions were quenched with 90 μL 25 mM succinic acid, 25 mM citric acid (pH 2.4), and cooled rapidly to 0 °C. Proteins were digested by adding 10 μL pepsin (4 μg) and analyzed immediately by LC-MS. Time-zero measurements were performed by quenching the reaction before adding D2O.
Peptides in pepsin digests of ERK1 were identified by LC-MS/MS, analyzing replicate digests on two instruments in order to maximize the probability that a peptide would be selected for MS/MS. Aliquots (4 μg) of proteolyzed ERK1 were analyzed on the QStar mass spectrometer, with m/z window 400–1600 Da, duty cycle 15.5 s, and three MS/MS per cycle. Aliquots (60 ng) of ERK1 were analyzed on an LTQ-Orbitrap mass spectrometer interfaced with an Eksigent 2DLC HPLC (75 μm i.d.× 150 mm column, Zorbax C18 resin), with m/z window 300–2000 Da, duty cycle 4-6 s (10–14 cycles/min), and five MS/MS per cycle. MS/MS were converted to .mgf files and searched against the human ERK1 sequence using MASCOT (v. 1.9), with no enzyme specified. Mass tolerances were 2.5 Da (parent) and 1.2 Da (fragment) for QStar datasets, and 1.2 Da (parent) and 0.8 Da (fragment) for LTQ-Orbitrap datasets. Peptides with MOWSE score greater than 20 were validated by manual analysis of MS/MS spectra. LC-MS of ERK1 was then carried out in water on the QStar, and only peptides with validated MS/MS that were also observed in the LC-MS run were reported.
HX-MS measurements of protection by Mg+2-AMP-PNP were carried out as above, with the following modifications. Prior to the HX reaction, 8 μL (5 μg) of 0P-ERK1 or 2P-ERK1 was incubated on ice for 10 min with either 7 μL of 21 mM MgCl2 + 2.1 mM AMP-PNP or 7 μL of 21 mM MgCl2. At t=0, exchange was initiated by adding 85 μL of a D2O/solute mixture to the kinase solution to reach a final buffer composition of 85% (v/v) D2O, 25 mM KPO4 (pH 7.4), 50 mM KCl, and 0.25 mM DTT, 10 mM MgCl2, and ± 1 mM AMP-PNP. Samples were incubated between 8 s and 110 min at 10 °C to allow in-exchange, then quenched by acidification with 90 μL 25 mM sodium succinate, 25 mM sodium citrate (pH 2.4), and cooled to 0 °C. Proteins were digested by addition of pepsin, and peptides were analyzed on the QStar mass spectrometer. Time-zero controls were performed by quenching the sample prior to addition of the D2O/solute mixture. To avoid systematic variations, time points and the order of sample analysis ± AMP-PNP were randomized.
Weighted average mass measurements were facilitated by using in-house HX-Analyzer software [15]. Samples were randomized to control for systematic variations, and replicate runs were performed at 0 s and 60 s in order to quantify the average standard deviation of weighted average mass (0.05 Da) and maximum standard deviation (0.13 Da). Estimates of artifactual in-exchange and back-exchange were obtained as described [18,19]. Back-exchange was estimated in two ways. First, back-exchange was calculated for each peptide using an empirical formula which assumes 1% back-exchange per minute after quenching [Eqn. (4) in reference 18]. Second, back-exchange was estimated by desalting and then lyophilizing peptides generated from pepsin digestion of 2P-ERK1, incubating with 90% D2O for 90 min at 90 °C to allow complete in-exchange, and measuring peptide mass by LC-MS under HX experimental conditions [18,20]. When counting the number of exchangeable amides for a given peptide, we included the first amide proton and did not assume rapid back-exchange on the timescale of the experiment. The two methods for estimating back-exchange were in reasonable agreement, where the measured values yielded an average of 28 ± 7 % back-exchange for 21 observable peptides, while the empirical calculation yielded an average of 22 ± 2 % back-exchange for the same peptides. Therefore we applied the second method involving back-exchange calculations to all peptides.
After correcting weighted average masses for artifactual in-exchange and back-exchange, deuteration time courses were fitted by nonlinear least squares to the equation Y = N − Ae−k1t − Be−k2t − Ce−k3t, where Y is the number of deuterons exchanged at time t; A, B, and C are the numbers of backbone amides exchanging at rates k1, k2, and k3, respectively; and N is the maximal deuteration over the experimental time period (N=A+B+C). Subtracting N from the total number of exchangeable backbone amides yields NE, the number of amides that are non-exchanging over the experimental time period. Nonlinear least squares calculations and graphics of time courses were generated using Sigmaplot v9.0 (SPSS Inc.).
Results
Hydrogen exchange measurements
Hydrogen-deuterium in-exchange measurements of ERK1 were obtained by LC-MS as described in Materials and Methods. Fifty-five peptides were identified after pepsin proteolysis, ranging in length between 6 and 36 residues (Table S1). These accounted for 98 % coverage of the primary sequence (370 of 379 residues), and 94 % coverage of exchangeable amides (334 of 356 amides). The peptides are annotated onto the primary sequence of His6-ERK1 in Fig. 1.
Figure 1. Pepsin cleavage map of human ERK1.

Shown is the sequence of His6-ERK1, indicating residue numbers and secondary structure as reported by Kinoshita et al. [5]. All peptides indicated were present in LC-MS/MS datasets of ERK1 in water, as well as HX datasets in D2O. Peptides indicated by stippled bars (1-29, 188-203, and 204-209) were observed only in 0P-ERK1 datasets, and the peptide indicated with horizontal stripes (188-209) was observed only in 2P-ERK1 datasets, reflecting differences in proteolysis in the activation lip. One peptide indicated by a solid bar showed increased in-exchange following phosphorylation and activation, and peptides indicated by dotted fill showed decreased in-exchange following phosphorylation and activation (see time courses in Suppl. Fig. 2).
Information about the degree of HX occurring within localized regions of 0P-ERK1 was assessed by measuring the extent of deuteration into different peptides over the 4 h timescale of the experiment, and mapping these against an X-ray structure determined for ERK1 [5] (Fig. 2A, Table S2). Slow exchange, leading to deuteration of less than 25% of exchangeable amides, was observed within core regions of the kinase corresponding to helix αE and strand β6 within the C-terminal lobe, the latter which contains the catalytic base within the active site. Slow exchange was also observed in the loop between helix αC and β4 as well as in strand β5, which contain residues that create a hydrophobic core within the N-terminal lobe. In ERK2, these residues form intramolecular connectivities with distal residues that in turn prevent autophosphorylation and autoactivation of the enzyme by constraining flexibility at the activation lip [21]. Fast exchange, leading to >75% deuteration of exchangeable amides, was observed within loop regions, including part of the activation lip and loops connecting the MAP kinase insert with C-terminal helices αG and αH. Deuteration exceeding 50% of exchangeable amides was also observed within N-terminal loop regions, the P+1 substrate binding loop, and the MAP kinase insert.
Figure 2. Hydrogen exchange patterns in ERK1.
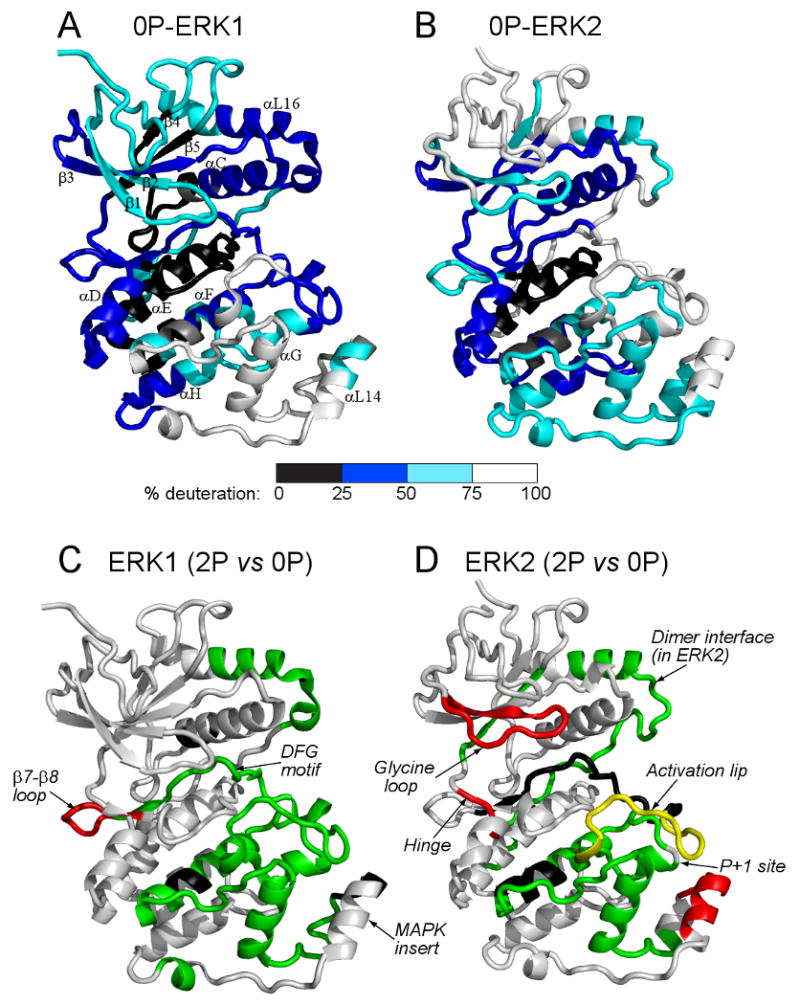
(A,B) Extent of deuteration into (A) 0P-ERK1 and (B) 0P-ERK2 after 4 h, normalized by the number of exchangeable amides in each region. For reach region, the extent of deuteration was measured as the sum of A+B+C (Suppl. Table 2), and mapped as colors corresponding to 0-25% (Black), 25-50% (Blue), 50-75% (Cyan) and >75% (White) of exchangeable amides. Overall, ERK1 and ERK2 showed similar patterns of regional hydrogen exchange, with differences that could be attributed to variations in sequence or structure. (C,D) Changes in HX induced upon phosphorylation and activation of (C) ERK1 and (D) ERK2. Peptides that showed increased (red) or decreased (green) HX following activation by phosphorylation were colored against the X-ray structures of 1P-ERK1 (2ZOQ) and 0P-ERK2 (1ERK). Peptides found in only one form of ERK1 or ERK2 (black) precluded comparison between inactive and active kinases. Yellow denotes a peptide in ERK2 which showed both increased and decreased HX rates upon activation. Of particular interest was the hinge region, which in ERK2 underwent enhanced HX surrounding the pivot point for interdomain closure, but in ERK1 was unaltered by kinase activation (see Suppl. Fig. 3). Data on ERK2 were from Lee et al. [11].
The extent of deuteration in ERK1 revealed many similarities with previous HX measurements of ERK2 [9,11] (Fig. 2A,B). Both enzymes showed low levels of exchange within the core regions of the N- and C-terminal domains, including the active site, helices αE and αF in the C-terminal domain, and the N-terminal core containing β3–αC–β4–β5. In both enzymes, high levels of exchange occurred within loops and peripheral secondary structure including the N-terminal strands β1LO-β2LO and β1-β2, intervening loop regions between β4-β5 and αF-αG, and the MAPK insert. Interestingly, some dissimilarities in HX behavior were observed between ERK1 and ERK2. Several prominent regions that underwent nearly complete in-exchange in ERK2 showed only partial exchange in ERK1; these included the activation lip and C-terminal helix αL16, which showed less than 50% exchange in ERK1, but greater than 90% exchange in ERK2. These differences in HX suggested lower conformational mobility in ERK1 compared to ERK2 within regions of the molecule linked to catalytic function.
Regulation of HX by ERK1 activation
Time courses comparing 0P-ERK1 to 2P-ERK1 (Suppl. Fig. 2) revealed significant changes in hydrogen exchange behavior following activation of ERK1 by dual phosphorylation. Sites of proteolytic cleavage were nearly identical between the 0P and 2P forms, with the exception of the activation lip, where cleavage between residues Glu203-Tyr204 (numbered as in Fig. 1) was observed only in the unphosphorylated form of ERK1. In total, eight peptides exhibited altered in-exchange behavior between 0P-ERK1 and 2P-ERK1 (Fig. 1, Figs. 3A-H). In 7 of 8 cases, kinase activation led to decreased HX rates; these included peptide 180-187 (LKICDFGL), containing the conserved DFG motif; peptide 210-216 (YRAPEIM), containing the P+1 substrate binding loop; peptides 218-232 (NSKGYTKSIDIWSVG) and 236-254 (AEMLSNRPIFPGKHYLDQL), containing αF-αG; peptides 255-271 (NHILGILGSPSQEDLNC) and 280-306 (YLQSLPSKTKVAWAKLFPKSDSKALDL), containing the αL14 MAP kinase insert and αH; and peptide 349-365 (AMELDDLPKERLKIELIF), containing the C-terminal helix αL16. These could either reflect structural changes leading to solvent inaccessibility, or else decreased flexibility and internal motions, following activation lip phosphorylation. Only one peptide exhibited increased HX upon activation; this region corresponded to the β7–β8 loop (peptide 173-180, LINTTCDL), which forms part of a docking motif binding site (Fig. 3A), and might reflect increased backbone flexibility within a binding pocket for high affinity interactions with substrates, MKKs, and other effectors.
Figure 3. HX time courses for peptides in ERK1 altered upon kinase activation.
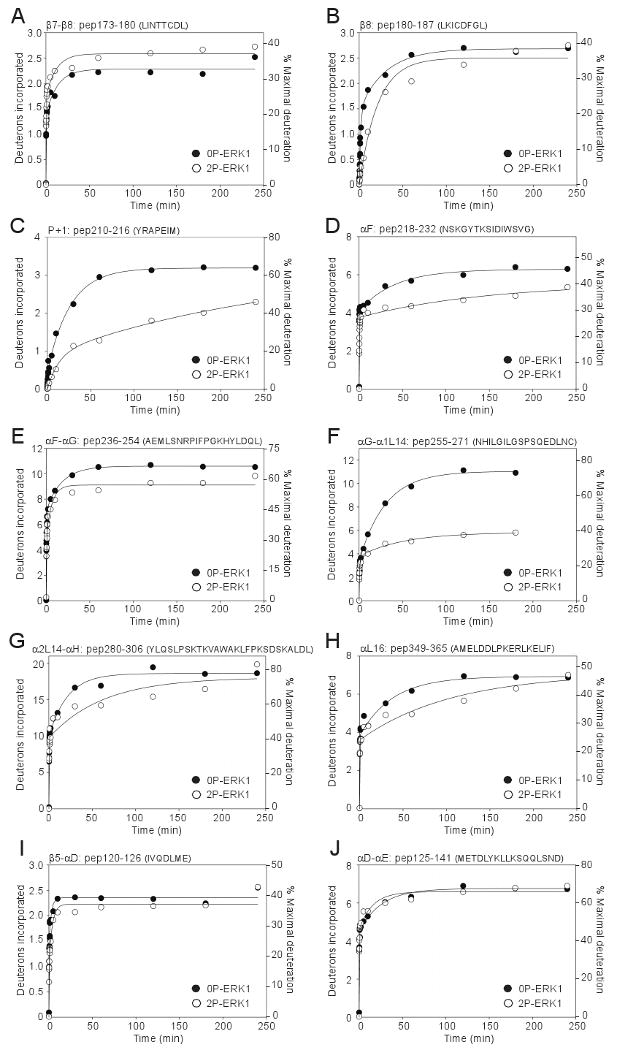
(A-J) Peptides are labeled by residue numbers as in Table S1, followed by the corresponding amino acid sequence. HX data on 0P-ERK1 are indicated by closed circles (●), and data on 2P-ERK1 are indicated by open circles (○). (I,J) Peptides 120-126 and 125-141 include the hinge region which in ERK1 showed no change upon activation, although HX within the sequence METDL was increased upon activation, in ERK2 [9,11].
The localized regions which showed HX alterations upon kinase activation were mapped against the backbone structure of ERK1, and compared to those previously reported for ERK2 (Figs. 2C,D). ERK1 and ERK2 showed some similarities in the patterns of HX that were correlated with activity. Both enzymes showed activation-induced changes in HX within the activation lip, a region which undergoes significant conformational remodeling in ERK2, which would also be expected to occur in ERK1. Decreased HX was observed in both enzymes at the C-terminal αL16 helix and surrounding regions, which in ERK2 has been shown to form an interface for dimerization. Similarly, decreased HX was observed in both enzymes within the P+1 substrate interaction loop, as well as helices αF and αG, which comprise the likely interface for extended substrate binding interactions. ERK2 showed few structural changes within this region that might account for the observed changes in HX, suggesting that reduced hydrogen exchange rates reflected decreased conformational mobility following kinase activation. Decreased HX had also been observed in this region following activation of p38α MAP kinase [15], suggesting that decreased flexibility in the substrate binding interface is a feature common to several MAP kinases.
ERK1 and ERK2 also showed key differences in their patterns of HX regulated by activation. For example, ERK1 showed no increased HX in the MAP kinase insert, which had previously been observed in ERK2 and ascribed to disrupted hydrogen bonding with activation lip residues upon kinase activation [9]. Likewise, ERK1 did not recapitulate the increased HX observed in the glycine-rich loop upon activation of ERK2, a region implicated in nucleotide binding. Importantly, differences were noted between the two enzymes in the hinge region localized between the N- and C-terminal domains, which in cAMP-dependent protein kinase corresponds to the pivot point for rigid body rotation and interdomain closure needed for formation of the catalytic site. In ERK2, this region of the hinge (METDL, Suppl. Fig. 3) showed significant enhancement of HX upon activation, which correlated with altered internal motions, allowing the kinase to switch from an open to closed solution conformation [9-11]. In contrast, peptide 125-141 (METDLYKLLKSQQLSND), peptide 125-145 (METDLYKLLKSQQLSNDHICY), and the overlapping peptide 130-145 (YKLLKSQQLSNDHICY) exhibited no change in HX behavior between active and inactive states in ERK1, from which we conclude that no change in HX occurs within the corresponding METDL hinge sequence in ERK1 (Fig. 3I,J, Suppl. Fig. 3). This suggested that hinge flexibility is not regulated by activation of ERK1 as it is in ERK2, and that the two enzymes differ with respect to the regulation of interdomain closure.
Domain closure in ERK1 can be monitored by HX-MS
The results above suggested that unlike ERK2, the hinge flexibility of ERK1 might not be controlled by activity state, and thus might allow interdomain closure in both inactive and active forms. In order to test this hypothesis, we conducted an HX-MS experiment which measured the steric protection from HX upon AMP-PNP binding to ERK1. Prior studies with ERK2 have shown that AMP-PNP protects N-terminal and hinge regions in the inactive and the active kinase, as expected from the known interactions between kinases and ATP, and that active ERK2 shows additional protection of the conserved DFG motif in the C-terminal domain. Thus, steric protection of the DFG motif reflects interdomain closure between N- and C-terminal lobes in solution. If our hypothesis were correct, we would expect to see protection of the DFG motif comparable in both forms of ERK1.
We measured HX time courses in the presence or absence of 1 mM Mg2+-AMP-PNP, comparing inactive vs active ERK1 (Suppl. Fig. 4). Only twenty of the 54 peptides were observed in these experiments between each of the four conditions, which nevertheless included most of the ERK1 sequence. Of these, four peptides showed significant reduction in HX rates upon AMP-PNP binding (Fig. 4A-D). Three of these corresponded to regions which are known to form contacts with nucleotide within the N-terminal domain. These included peptide 46-69 (QYIGEGAYGMVSSA), containing the glycine loop which forms hydrogen bond interactions with β and γ-phosphate groups of ATP and forms a flexible gate for nucleotide binding; peptide 60-88 (YDHVRKTRVAIKKISPFEHQTYCQRTLRE), containing conserved Lys and Glu residues in strand β3 and helix αC, respectively, which form ion-pair interactions with ATP α and β-phosphate groups; and peptide 115-124 (MRDVYIVQDL), containing residues in strand β5 which form hydrogen bonding interactions with nucleotide (Fig. 4A-C). The fourth peptide, peptide 181-187 (KICDFGL), contains the conserved DFG motif (Fig. 4D). Here, we observed comparable protection upon AMP-PNP binding to both inactive and active kinase forms. This was in contrast to the corresponding DFG region in ERK2, where previous measurements revealed 10-fold greater nucleotide protection from HX in the active kinase compared to the inactive form (Suppl. Fig. 5) [11]. Our measurements of ERK1 indicate that that nucleotide interacts productively with both the N-and C-terminal domains in 0P- and 2P-ERK1, and more productively with the C-terminal domain in 0P-ERK1 than 0P-ERK2. Taken together, the findings suggest that both inactive and active forms of ERK1 are able to adopt interdomain interactions leading to a closed conformation in solution.
Figure 4. HX time courses for peptides showing protection of ERK1 upon nucleotide binding.
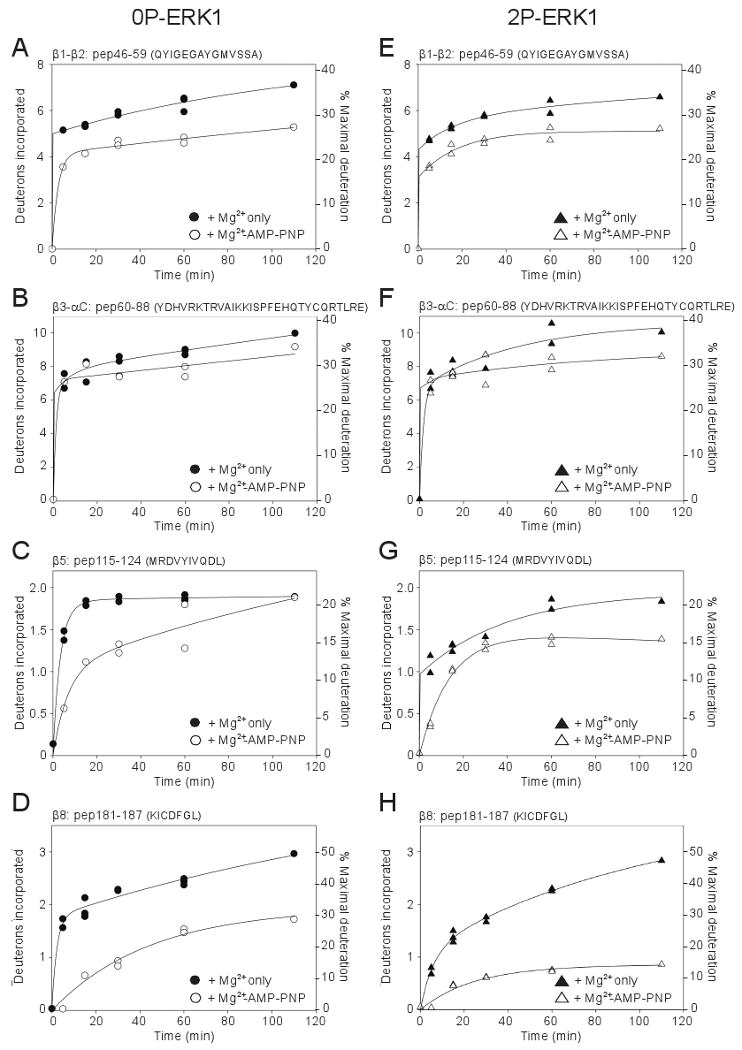
(A-H) Peptides from 0P-ERK1 (left panels) and 2P-ERK1 (right panels) show effects of AMP-PNP binding to ERK1. HX data on apoenzyme were indicated by closed symbols, and data collected with AMP-PNP were indicated by open symbols. Both 0P-ERK1 and 2P-ERK1 showed significant protection of HX upon binding AMP-PNP, suggesting interdomain closure in both inactive and active forms.
Discussion
This study reveals novel insight into the manner in which internal motions in ERK1 are regulated by enzyme activation, as revealed by HX-MS. We demonstrate that ERK1 and ERK2, which are 90% identical in primary sequence, show differences in HX behavior that suggest differences in conformational mobility between closely related kinases.
Interpretation of HX results can be greatly aided by the availability of information on atomic structure. Numerous X-ray structures of ERK2 have been solved in varying forms, including unphosphorylated, diphosphorylated, peptide-bound, inhibitor-bound, or harboring mutations within the phosphorylation lip and active site. In contrast, only one X-ray structure of ERK1 has been published to date, representing a form which is mono-phosphorylated at its regulatory Tyr residue (1P-ERK1, ref. 5), and whose activity is approximately 10% of the fully active, diphosphorylated enzyme [17]. The tertiary structure of ERK1 shows a consensus kinase core structure containing two insertions unique to MAP kinases (αLO, αL14). The backbone conformation is very similar to the structure of ERK2 [6-8], consistent with the extensive sequence identity between these enzymes. In its configuration of the activation lip, the 1P-ERK1 structure appears most consistent with what would be expected for an inactive conformation of the enzyme, resembling the inactive, unphosphorylated form of ERK2 (0P-ERK2) more than the active diphosphorylated form (2P-ERK2).
While the overall extent of HX was generally similar between 0P-ERK1 and 0P-ERK2 and consistent with the conserved tertiary structures, certain dissimilarities were observed. For example, the extent of HX was lower in ERK1 than ERK2 within the β1LO-β2LO and β1 strands of the N-terminal lobe, and higher in regions surrounding the MAP kinase insertion. Both of these regions showed significant differences in primary sequence as well as structure (Suppl. Fig. S6 sequence alignment). Likewise, HX was lower in ERK1 within the β7-β8 loop and the C-terminal αL16 helix, suggesting lower flexibility. Both regions showed similar sequences, but higher structural order in ERK1 than ERK2, reflected by closer beta-sheet hydrogen bonding distances in the β7-β8 loop of ERK1, as well as an additional helical turn proximal to αL16 in ERK1 which was absent in ERK2. Lower HX was also observed around the activation lip of ERK1, compared to ERK2. Although primary sequences and conformations were similar between these regions of ERK1 and ERK2, residues immediately adjacent to pTyr as well as within helix αC in ERK1 were distorted relative to 0P-ERK2 in a manner suggesting greater similarity to corresponding regions in 2P-ERK2, which might account for the partial activity of 1P-ERK1. Thus, for the most part, differences in extent of HX between ERK1 and ERK2 could be attributed to variations in sequence and/or structure.
Intriguing differences were observed between ERK1 and ERK2, when we assessed the effects of phosphorylation and activation on conformational mobility. Our previous studies on ERK2 showed that enzyme activation led to significant changes in HX within localized regions of the protein. These could be attributed to changes in protein flexibility and internal motions, especially when HX changes occured in a manner inconsistent with structural differences between 0P-ERK2 and 2P-ERK2. In particular, regions of ERK2 that showed regulated HX ascribed to changes in flexibility were those implicated in substrate binding or catalysis, which could conceivably contribute to enzyme activation. This was further examined in the hinge region, where increased flexibility upon kinase activation correlated with release of constraints to interdomain closure. In the case of ERK1, the limited X-ray information was insufficient to draw dependable conclusions about how well the changes in HX could be explained by changes in structure vs internal motions. Nevertheless, to the extent that ERK1 behaves similarly to ERK2, whose conformational changes were largely restricted to the activation lip and active site, our HX measurements on ERK1 could conceivably reflect effects of activation on protein dynamics. As in ERK2, the regions showing regulated HX in ERK1 are associated with kinase function. Increased HX in ERK1 upon activation may reflect increased mobility of the β7-β8 loop which is involved in substrate docking motif recognition. Likewise, reduced mobility upon activation might also occur in the DFG motif which is involved in ATP binding, in the C-terminal helix αL16, which is involved in kinase dimerization, and in the P+1 loop and helices αF-αG, which form the interface for substrate binding and recognition.
Notably, differences were observed between the two enzymes in the hinge region, which in ERK2 showed increased backbone flexibility upon activation, but in ERK1 showed little change between active and inactive states (Suppl. Fig. 3). Further evidence showing that AMP-PNP interacts productively with N- and C-terminal domains in both 0P-ERK1 and 2P-ERK1 suggests a model in which both the active and inactive states of ERK1 allow interdomain closure and formation of a closed active site (Fig. 5). This behavior differs substantively from ERK2, where constraints to domain closure consistent with an open solution conformation were observed in the inactive, unphosphorylated form, and were released upon activation by diphosphorylation, enabling the formation of a closed solution conformation that would be needed for catalysis. We conclude that ERK1 and ERK2, while closely related in sequence and structure, nevertheless differ significantly in their constraints to kinase activation, and the mechanisms by which they overcome these barriers through activation-induced regulation of conformational mobility.
Figure 5. Summary of results and conceptual model.
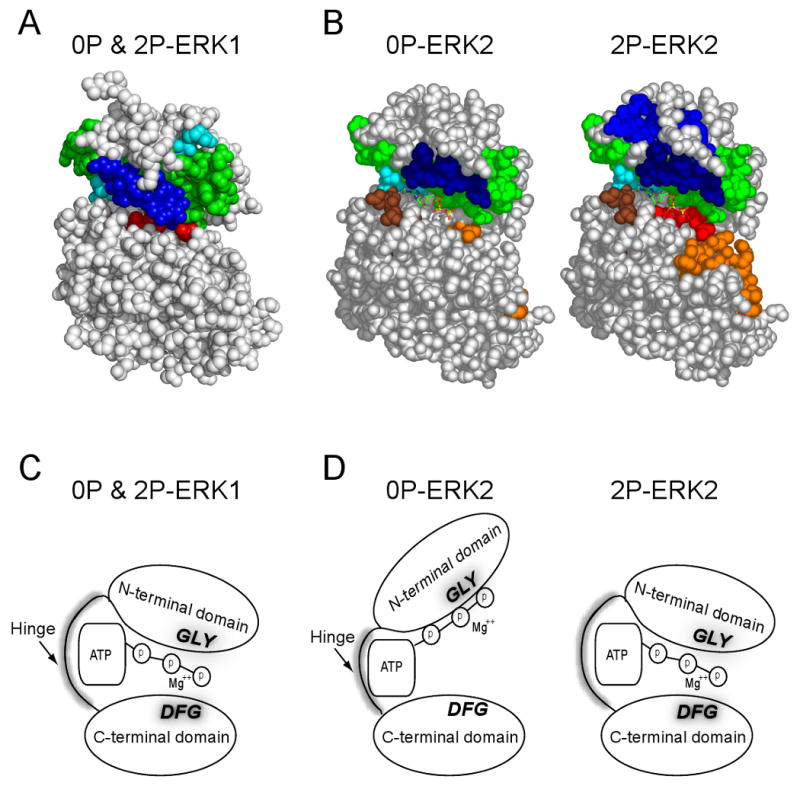
(A,B) Peptides protected from HX by Mg+2-AMP-PNP binding are shown in color. (C) Model of results, showing interdomain interactions leading to domain closure in both 0P- and 2P-ERK1, based on HX protection in the C-terminal domain. (D) Model indicating that in solution, interdomain interactions leading to domain closure are enhanced in ERK2 by phosphorylation and activation.
Research Highlights.
Hydrogen-exchange mass spectrometry (HX-MS) measures protein conformational mobility. Inactive vs active forms of the MAP kinase, ERK1, are compared by HX-MS. Kinase activation affects conformational mobility differently in related MAP kinases. Interdomain closure is constrained prior to activation of ERK2, but not ERK1. MAP kinases have distinct mechanisms for activation via control of protein motions.
Supplementary Material
Acknowledgments
We are indebted to Stephane Houel, Shuji Kato, Kutralanathan Renganathan, and William Old for assistance with LC-MS/MS and peptide search programs, to Rebecca West for help with data analysis, and to Melanie Cobb for her gift of ERK1 expression plasmids. This work was supported by NIH grant GM074134 (NGA) and a fellowship for undergraduate independent research from the Undergraduate Research Opportunities Program (UROP) at the University of Colorado, Boulder (AYR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baselga J. Targeting tyrosine kinases in cancer: the second wave. Science. 2006;312:1175–1178. doi: 10.1126/science.1125951. [DOI] [PubMed] [Google Scholar]
- 2.Montagut CJ, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283:125–134. doi: 10.1016/j.canlet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Pagès G, Guérin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouysségur J. Defective thymocyte maturation in p44 MAP kinase (Erk1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- 4.Yao Y, Li W, Wu J, Germann UA, Su MS, Kuida K, Boucher DM. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci U S A. 2003;100:12759–12764. doi: 10.1073/pnas.2134254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinoshita T, Yoshida I, Nakae S, Okita K, Gouda M, Matsubara M, Yokota K, Ishiguro H, Tada T. Crystal structure of human mono-phosphorylated ERK1 at Tyr204. Biochem Biophys Res Commun. 2008;377:1123–1127. doi: 10.1016/j.bbrc.2008.10.127. [DOI] [PubMed] [Google Scholar]
- 6.Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 7.Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 8.Fox T, Coll JT, Xie X, Ford PJ, Germann UA, Porter MD, Pazhanisamy S, Fleming MA, Galullo V, Su MS, Wilson KP. A single amino acid substitution makes ERK2 susceptible to pyridinyl imidazole inhibitors of p38 MAP kinase. Protein Sci. 1998;7:2249–2255. doi: 10.1002/pro.5560071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoofnagle AN, Resing KA, Goldsmith EJ, Ahn NG. Changes in protein conformational mobility upon activation of ERK2, as detected by hydrogen exchange. Proc Natl Acad Sci USA. 2001;98:956–961. doi: 10.1073/pnas.98.3.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoofnagle AN, Stoner JW, Lee T, Eaton SS, Ahn NG. Phosphorylation-dependent changes in structure and dynamics in ERK2 detected by site directed spin labelling and electron paramagnetic resonance. Biophys J. 2004;86:395–403. doi: 10.1016/S0006-3495(04)74115-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee T, Hoofnagle AN, Resing KA, Ahn NG. Hydrogen exchange solvent protection by an ATP analogue reveals conformational changes in ERK2 upon activation. J Mol Biol. 2005;353:600–612. doi: 10.1016/j.jmb.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Harkins PD, Ulevitch RJ, Han J, Cobb MH, Goldsmith EJ. The structure of mitogen-activated protein kinase p38 at 2.1 Ǻ resolution. Proc Natl Acad Sci USA. 1997;94:2327–2332. doi: 10.1073/pnas.94.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson KP, Fitzgibbon MJ, Caron PR, Griffith JP, Chen W, McCaffrey PG, Chambers SP, Su MS. Crystal structure of p38 mitogen-activated protein kinase. J Biol Chem. 1996;271:27696–27700. doi: 10.1074/jbc.271.44.27696. [DOI] [PubMed] [Google Scholar]
- 14.Bellon S, Fitzgibbon MJ, Fox T, Hsiao HM, Wilson KP. The structure of phosphorylated p38 gamma is monomeric and reveals a conserved activation-loop conformation. Structure. 1999;7:1057–1065. doi: 10.1016/s0969-2126(99)80173-7. [DOI] [PubMed] [Google Scholar]
- 15.Sours KM, Kwok SC, Rachidi T, Lee T, Ring A, Hoofnagle AN, Resing KA, Ahn NG. Hydrogen exchange mass spectrometry reveals activation-induced changes in conformational mobility of p38α MAP kinase. J Mol Biol. 2008;379(5):1075–1093. doi: 10.1016/j.jmb.2008.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilsbacher JL, Cobb MH. Bacterial expression of activated mitogen-activated protein kinases. Methods Enzymol. 2001;332:387–400. doi: 10.1016/s0076-6879(01)32217-6. [DOI] [PubMed] [Google Scholar]
- 17.Robbins DJ, Zhen E, Owaki H, Vanderbilt CA, Ebert D, Geppert TD, Cobb MH. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- 18.Resing KA, Hoofnagle AN, Ahn NG. Modeling deuterium exchange behavior of ERK2 using pepsin mapping to probe secondary structure. J Am Soc Mass Spec. 1999;10:685–702. doi: 10.1016/S1044-0305(99)00037-9. [DOI] [PubMed] [Google Scholar]
- 19.Lee T, Hoofnagle AN, Resing KA, Ahn NG. Protein hydrogen exchange measured by electrospray ionization mass spectrometry. In: Celis JE, editor. Cell Biology: A Laboratory Handbook. Third. Vol. 4. Elsevier Science; 2006. pp. 443–449. [Google Scholar]
- 20.Resing KA, Ahn NG. Deuterium exchange mass spectrometry as a probe of protein kinase activation. Analysis of wild-type and constitutively active mutants of MAP kinase kinase. Biochemistry. 1998;37:463–475. doi: 10.1021/bi971750x. [DOI] [PubMed] [Google Scholar]
- 21.Emrick MA, Lee T, Starkey P, Mumby MC, Resing KA, Ahn NG. The gatekeeper residue in ERK2 controls autoactivation via a pathway of intramolecular connectivity. Proc Natl Acad Sci USA. 2006;103:18101–18106. doi: 10.1073/pnas.0608849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.