




Abstract
Gene regulatory networks (GRNs) provide system level explanations of developmental and physiological functions in the terms of the genomic regulatory code. Depending on their developmental functions, GRNs differ in their degree of hierarchy, and also in the types of modular sub-circuit of which they are composed, although there is a commonly employed sub-circuit repertoire. Mathematical modelling of some types of GRN sub-circuit has deepened biological understanding of the functions they mediate. The structural organization of various kinds of GRN reflects their roles in the life process, and causally illuminates both developmental and evolutionary process.
The body plan of an animal, and hence its exact mode of development, is a property of its species and is thus encoded in the genome. Embryonic development is an enormous informational transaction, in which DNA sequence data generate and guide the system-wide spatial deployment of specific cellular functions. GRNs also determine the main events of postembryonic development, including organogenesis and formation of adult parts and cell types. Beyond that, GRNs control a vast array of physiological capabilities and modes of response to environmental fluctuations and challenges. GRNs are composed of multiple sub-circuits, that is, the individual regulatory tasks into which a process can be parsed are each accomplished by a given GRN sub-circuit1–4. Thus the operational significance of a GRN structure will be indicated by the types of sub-circuit it contains. However, GRNs have more global organizational properties as well. The comparative review below shows that GRNs may be deeply layered, generating successive regulatory transactions, or they may be shallow, in the sense that they mandate few transactions between the initial inputs and the terminal activation of effector genes.
The developmental GRN sub-circuit repertoire
Modular GRN sub-circuits are defined by their topologies, and the topology of a sub-circuit directly indicates its function in life. In this article I am concerned only with sub-circuits which perform developmental biology jobs that can be defined uniquely, and not with very common ‘motifs’ such as the coherent feed forward loop, which although it has specific dynamic properties5, appears in so many different contexts that no unique developmental biology function can be associated with it. Table 1 contains a compilation of sub-circuits drawn from all the various GRNs considered in this review, together with an abbreviated description of their regulatory functions, and abbreviated diagrams illustrating the canonical sub-circuit structures. Additional sub-circuits will be found as more developmental GRNs are explored, but the basic import of Table 1 is that there probably exists a small, finite number of sub-circuit topologies out of which developmental programs of all kinds are constructed. The first entry, for example, is a spatial information processing sub-circuit called the double-negative gate, found in the sea urchin embryo GRNs2,6. This sub-circuit consists of two genes encoding repressors wired in tandem, so that the target of the first repressor is the gene encoding the second, plus downstream regulatory genes which are targets of the second repressor. Its function is to ensure that the target genes are expressed only where the first repressor is (transiently) active (domain X), while these genes are shut down everywhere else (1 − X); what we term an X,1 − X processor.
Table 1.
Sub-circuit repertoire for developmental GRNs
| Regulatory state specification function |
Sub-circuits | What they do | Topologies | |
|---|---|---|---|---|
| X,1 − X processors | Double negative gate1,2,6 | Install regulatory state in X domain, prohibit same state everywhere else* | 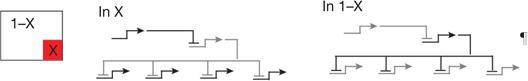 |
|
| 1.1 | ||||
| Signal-mediated switch2 | Activate regulatory gene(s) in cells receiving signal, repress same genes everywhere else† |  |
||
| 1.2 | ||||
| Spatial subdivision | Inductive signaling2 | Activation of new regulatory genes in a cellular domain by transcriptional response to signal ligands produced by other cells | 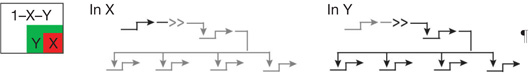 |
|
| 2.1 | ||||
| AND logic circuitry2 | Overlapping but spatially non-coincidental inputs are generated and both are required for regulatory gene activation, which occurs only in overlap subdomain | 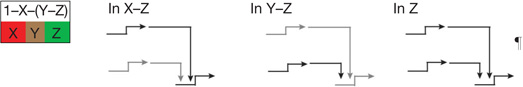 |
||
| 2.2 | ||||
| Spatial repression2 | Boundaries of spatial regulatory state domains controlled by transcriptional repression. | 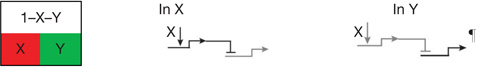 |
||
| 2.3 | ||||
| Dynamic lockdown of regulatory state‡ | Reciprocal repression of state1,2,9,39,51 | In each spatial regulatory state domain key activators of alternative states are transcriptionally repressed by ‘exclusion’ circuitry§. | 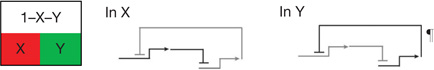 |
|
| 3.1 | ||||
| Feedback circuitry1 | Two or three regulatory genes engage in positive intergenic feedback, stabilizing regulatory state irrespective of transient inputs | 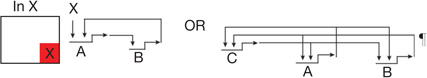 |
||
| 3.2 | ||||
| Community effect circuitry2,23,92 | Cells within a territory all signal to one another, driving continued uniform expression both of ligand gene and signal-dependent regulatory genes | | |  |
||
| 3.3 | ||||
| Boundary maintenance | Reciprocal repressive signalling across boundary32 | Different signals are produced by apposing cells and their reception triggers repressive circuitry excluding the cross-boundary regulatory state | 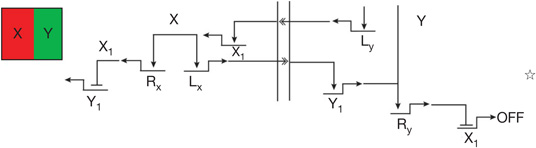 |
|
| 4 | ||||
| Terminal binary cell fate choice | Alternate sub-circuits driven by reciprocal repressors8,36–38,81 | External inputs tip the balance of repressor expression, resulting in activation of one differentiation program and exclusion of the other | 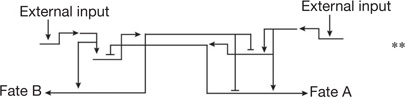 |
|
| 5 | ||||
| Discontinuous transcriptional response to signal intensity and/or duration | Reciprocal repressor genes responding cooperatively to inducer21,38,81 | Circuitry generates differential stimulation of expression of reciprocal repressors in low versus high signal intensity†† |  |
|
| 6.1 | ||||
| Reciprocal repressor genes, one activating an additional repressor gene, each with variable external positive inputs82 | Circuitry generates irreversible transitions, in stem cell regulatory state, off versus on in response to signals of different strength and duration | 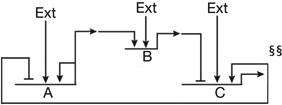 |
||
| 6.2 | ||||
| Triple feedback linkage with asymmetric signal inputs47 | Produces alternative regulatory states, or low level indeterminate state, depending of different positive inputs | | | | |  |
||
| 6.3 | ||||
The role of the sub-circuit is given in column 1; its name in column 2; a description of its function in column 3; and the sub-circuit structure in column 4. Numbers in column 2 are keyed to Fig. 1. See references indicated for actual occurrences, exact circuit topologies, and discussion of information processing specifics. In each case the functions of the circuit are hardwired in its cis-regulatory target sites. In Topologies column, all genes encode transcription factors unless otherwise noted.
Regulatory genes that create initial regulatory state are controlled by widely expressed repressor, which is dominant over their positive inputs, and gene encoding this repressor is itself specifically repressed in a local region (X) by another gene encoding a different repressor: hence target genes are ON in X, specifically repressed elsewhere.
Many developmental signalling systems (for example, Notch, Wnt) activate immediate early response factors in cells receiving ligand, but in absence of ligand, these factors act as dominant repressors of the same target genes.
Dynamic in that continuing transcription is required.
Exclusion sub-circuits are activated as downstream outputs of specification GRNs.
A unique circuit design here is that the ligand gene is activated by the same signal transduction mechanism reception of the ligand activates in recipient cells; a positive intercellular feedback.
From ref 2.
L, gene encoding signalling ligand.
R encodes repressor; L encodes signalling ligand.
This example was adapted from Ref 36.
Conceived as a means of obtaining different discrete transcriptional responses from a graded signal; see discussion of this type of circuitry in section on mathematical models below.
S, signal; triangle represents graded signal strength
S1, S2, different signal inputs gene B is subject to additional transcriptional repression in certain regulatory states.
This design precludes necessity for ad hoc Hill coefficients as in 5, 6.1; see section on mathematical models below.
Autoregulatory loops lock on whichever state the system goes to.
References in Table 1 generalize the point that structurally similar sub-circuits, but composed of different regulatory genes, are repeatedly encountered doing similar developmental jobs in different GRNs. At root this is because what the circuit can do depends directly on its structure; for example, in a recent study, a search of all possible small sub-circuits based on 3-node topologies showed that only two are capable of response to a signal followed by return to the original state7.
A given sub-circuit structure implies a given function, and in development there is a finite set of regulatory functions required. The biochemical complexity of the diverse cis-regulatory systems composing developmental GRN sub-circuits, and the diversity of the sets of transcription factors which animate them, thus may give way to a pleasingly simple set of logic-processing sub-circuit topologies. This would be a very important outcome, for it would make it possible to parse the apparently enormous mazes of interconnections in system level GRNs into modules of developmental logic function, and thus to understand how GRNs control the biology we see.
Deep structure of embryonic GRNs
The GRNs which control the de novo formation of embryonic territories typically include many different functional sub-circuits, which govern successive ‘layers’ of process. They are hierarchical in their overall structure. Their depth simply reflects the long sequence of regulatory steps required to complete any component of embryonic development. The concept of deep as opposed to shallow GRN structure can be simply considered as the number of successive changes in regulatory state required to generate an episode of embryological or other development, between the initial state and the terminal process which the GRN causes to happen. The terminal outcome is, by definition, the activation of cohorts of effector genes (that is, differentiation and cell biology genes, as opposed to only regulatory genes). In relatively shallow GRNs, some of which are considered below, the initial state may be a paused regulatory condition just upstream of expression of a differentiation gene battery.
The sea urchin embryo endomesoderm GRN serves as a reference point, as at present it is the most nearly complete, predictively useful, and validated large scale developmental GRN available. Structure/function aspects of this GRN have been reviewed recently2, and an always current version, together with underlying data and dynamic presentations by domain, is available at http://sugp.caltech.edu/endomes/. The concept of GRN depth is illustrated in Fig. 1a by abstracting from the sea urchin GRN the sequence of sub-circuits deployed in order to specify its skeletogenic cell lineage1, which produces only the one cell type, and is developmentally the simplest process modelled in the whole endomesoderm GRN. This portion of the network contains 24 regulatory genes and several signalling genes, as well as a sampling of downstream differentiation genes Without detailing the individual genes and linkages in the skeletogenic GRN, its internal structure is abstractly represented in Fig. 1a as a series of interconnected boxes, each of which represents a GRN sub-circuit that executes the indicated regulatory task. Many of these sub-circuits are among the types listed in Table 1, as indicated by the colour coding, and the arrows show the linkages from one sub-circuit to another, that is, they represent transcription factors generated in one box and used for control of gene(s) in the next box, the inputs or ‘feeds’ into each sub-circuit. The boxes are layered hierarchically, with those that initiate the process at the top. Figure 1a includes various control processes that are common throughout embryonic development, because the problems that have to be solved are general: the initial spatial inputs have to be interpreted, the regulatory state then has to be locked down (the initial inputs are always transient), signals have then to be generated, other states have to be excluded, and differentiation drivers have to be activated. It is not surprising that all this requires a lot of sequential circuitry, even given the relative simplicity of skeleto genic lineage development. The GRNs underlying specification of the mesoderm, endoderm1,2,8 and of the oral and aboral ectoderm9 of the sea urchin embryo are similarly deep, layered and hierarchical.
Figure 1. ‘Birdseye’ views of structural properties of representative developmental GRNs.

a–c, Diagrammatic view of sub-circuits and sub-circuit functions in three different GRNs. Each box represents a GRN sub-circuit consisting of a small number of regulatory genes and their functional linkages. Coloured dots and numbers refer to the similarly coded sub-circuit types in Table 1. Red arrows indicate linkages between sub-circuits, that is, regulatory feeds from one sub-circuit to another. a, GRN for skeletogenic mesoderm lineage specification in sea urchin embryos1. b, GRN for pancreatic developmental process3, leading to β cell specification and insulin gene transcription. c, GRNs typical of terminal binary fate choices in haematopoietic stem cells and other similar situations, as discussed in text.
GRNs for specification of mesoderm in Xenopus embryos10, for specification of the gut and mesoderm cell lineages in Caenorhabditis elegans embryos11, and for specification of endoderm and dorsal, anterior and ventral gene expression domains12, and of mesoderm13 in zebrafish, also display deep, hierarchical organizations. Some of these GRNs are compilations from the literature or from chromatin immunoprecipitation (ChIP)-chip observations, and have not been validated by direct perturbation analysis, let alone at the cis-regulatory level, but it is unlikely that their overall structure is illusory.
The GRN for dorsal/ventral patterning in Drosophila14, which does have extensive cis-regulatory support, is also hierarchical, but its unusual structure reflects the unusual developmental process it controls. In this embryo regulatory state domains are initially set up very quickly in a syncytium, without intercellular signalling, although following cellularization, signalling dominates further transcriptional functions and the GRN henceforth has a typical structure. In the syncytial embryo spatial stripes of both dorsal/ventral and anterior/posterior regulatory gene expression, which specify the future multicellular embryonic territories, are generated by parallel cis-regulatory responses to maternally localized and zygotically expressed combinations of diffusing transcription factors15–17. Many of these factors act as repressors in setting spatial boundaries. Important initial inputs in this system are the transcription factors Dorsal and Bicoid, encoded by maternal messenger RNAs which become distributed in graded fashion in the syncytial embryo nuclei, from ventral to dorsal and anterior to posterior, respectively. Cis-regulatory modules have been isolated that control target genes expressed in stripes at given ranges of values of these ‘morphogens’. When associated with reporters, and introduced into the egg, these cis-regulatory modules produce stripes of expression at the appropriate positions along the respective axes. It has been assumed for a long time that the positions where they operate are determined by the quantitative values of the Dorsal or Bicoid concentrations at those locations, which in some way these cis-regulatory systems read. Much recent evidence, however, shows that the positions where these cis-regulatory modules act depend on combinatorial activator and repressor inputs15–19, and the quantitative values of these ‘morphogens’ alone do not by themselves predict the spatial expression domains of their target genes (except sometimes at the extreme positions where their concentrations are highest). The combinatorial inputs are the products of regulatory genes linked into the GRN20. Thus the concept that the position of target gene expression is determined solely by the quantitative value of the ‘morphogen’ is overly simplistic: the overall pattern, and the overall signal strength response mechanism, are actually network properties rather than a property of individual cis-regulatory modules that independently and quantitatively read single gradient values.
Vertebrate embryos also use graded inputs, but in this case the developmental systems are cellular, and the ‘morphogens’ are diffusible extracellular signal ligands. Distinct regulatory responses occur, dependent on the intensity of signalling, resulting in activation of different genes in different locations in the embryo, for example in response to an activin gradient in the pre-gastrular Xenopus embryo21. Modelling shows that this particular level-sensitive response could be mediated by a specific type of GRN sub-circuit, in which regulatory genes encoding repressors reciprocally damp each other’s expression, while responding differentially in a cooperative, thus nonlinear and discontinuous way, to the concentration of the signal (Table 1). However, as we see in the following there is more than one type of sub-circuit capable of discontinuous response to a graded signal.
Structure of GRNs encoding body parts
We now have bits and pieces of the GRNs controlling development of body parts and organs in later embryonic development, usually their initial stages. Like embryonic GRNs, they are deep and hierarchical, and indeed are in most ways structured similarly to the early embryo GRNs. The box diagram cartoon in Fig. 1b provides an example. This diagram is abstracted from a GRN for specification of pancreas and then of pancreatic β-cells3. In adult body part formation, including organogenesis, the first step is always establishment of a given regulatory state in the field of cells from which the body part will form, the progenitor field22, for example, the cardiac crescent, or the limb bud, or the imaginal disc. The progenitor field is positioned with respect to the coordinates of the developing organism, which always involves signal-mediated installation of a new regulatory state. But then the field is subdivided into the regulatory state domains of its subparts, and at each step the state is locked down. This used to be called ‘pattern formation’, when people were looking at only one or a few genes at a time. As in early embryo GRNs the main job in setting up the parts and future form of the organ is the progressive deployment of regulatory states in space. It is essential to realize that this process is not to be equated with terminal cell fate specification; the cells expressing patterned regulatory states are yet far upstream in the developmental process from their ultimate descendants, which will eventually differentiate in various directions, according to what part of the organ they arise in.
The similarity between GRNs encoding adult body part formation and those controlling earlier embryogenesis is also sustained at the sub-circuit level in that few additional types of sub-circuit are used. For instance, in pre-gastrular embryonic specification it can be confidently predicted that a feedback circuit locking two or three regulatory genes in a mutual positive embrace will be encountered just downstream of the initial inputs used to set up a given regulatory state (Table 1). This is seen at multiple locations in the sea urchin embryonic GRNs1–3,23, and the same feature routinely appears in adult body part GRNs: for example, in those underlying development of neural crest in lamprey24, gut specification in vertebrates25, eye lens field specification in both vertebrates26 and Drosophila27, haemangioblast specification in mouse28, pharynx specification in C. elegans29, heart specification in mammals and Drosophila3,30,31, and pancreatic β-cell specification in mouse3. Each of these GRNs include two- or three-gene positive feedback sub-circuits functioning to lock down newly installed spatial regulatory states. The similar feedback sub-circuits are constructed with different genes; again it is the sub-circuit topology that determines function, and many different regulatory genes can have the same roles. An additional type of sub-circuit that is commonly seen in later embryonic processes is a signal-mediated, mutual repression device that operates across a cellular boundary, such that, on either side, reception of a signal from the other side specifically causes repression of key genes of the alternate regulatory state (Table 1). Four examples from later development where this type of circuitry obtains are in the GRN that maintains the distinct regulatory states of anterior and posterior parasegment compartments in Drosophila32, the GRN controlling establishment of dorsal/ventral neural tube domains in vertebrates33, the GRN controlling anterior versus posterior specification in the vertebrate limb bud34, and the GRN controlling cell type specification under signal control in the C. elegans vulva35.
Postembryonic developmental GRNs: differentiation from pluripotent stem cells
A remarkably recurrent similarity in GRN circuit design has recently emerged in studies of the transcriptional pathways that control binary fate choices executed in the diversification of haematopoietic cell types from multipotent precursors (for reviews, see refs 36–38). At the cores of these circuits, which use some overlapping and some lineage-specific regulatory genes, are pairs of genes encoding transcription factors that mutually antagonize each other’s expression within the same nucleus. Often initially co-expressed at relatively low levels, the lineage fate choice depends on stepped up asymmetric expression of one or the other of the core repressor gene pair. Each of these genes also directly or indirectly promotes expression of positive regulators necessary for execution of one of the lineage fate choices. As the activity of one of the core repressors increases, it causes transcriptional extinction of expression of the alternative choice, and the irreversible installation of its own positive regulatory state (see discussion of the mathematical features of such circuits below). An important point is that the genes of the antagonistic repressor pairs, and/or the regulatory genes that are their immediate targets, also provide direct positive or negative inputs into terminal differentiation genes of the alternate lineages37,39. In other words, this apparatus is deployed immediately upstream of the drivers of the effector genes that generate the features of given cell types (Fig. 1c). In comparison to the embryonic GRNs just considered, these are relatively shallow networks. Ultimately the decisive inputs into one or the other of the core repressors are provided by extrinsic signalling ligands, for example cytokines and growth factors, including Notch and Tgfβ, or endogenous immune receptor signals. The binary choice transcriptional apparatus responds to signal intensity, so that a low input gives one result and a high input another. Different pairs of repressor genes perform similar roles in different lineage fate choices, but what is remarkable is the similar circuitry adduced throughout haematopoietic diversification. Transcriptional balance between pairs of cross-antagonistic repressors decides the outcome, for instance, in myeloid progenitors giving rise to macrophages or neutrophils37; in precursors that may give rise to either B cells or macrophages38, where there is cis-regulatory evidence of the transcriptional cross-repression; in the upper level decision point where erythroid versus myeloid fates bifurcate40–42; in the erythroid versus platelet fate decision43. Similarly, in T-cell diversification between helper vs killer fate44,45, T-cell receptor signal strength indirectly controls repressor function, a compelling case because there is direct cis-regulatory evidence of the reciprocal transcriptional silencing interactions46.
Although to some it is tempting to view all development through the same lens, there are fundamental differences between the terminal fate choice circuitry discussed here and the GRNs that execute early and mid-stage embryonic development of animal body parts. Differentiation gene batteries can be activated only at the end of the series of GRN transactions that decide exactly where they are to be deployed. Haematopoietic cell fate decisions occur at the end of a complex prior developmental process, and in fact as discussed below, the circuitry controlling very early haematopoietic stem cell pluripotentiality operates in an entirely different manner from the binary choice circuitry just considered28,47. In their function, haematopoietic binary choice sub-circuits are similar to the terminal sub-circuits that elsewhere in development immediately determine deployment of differentiation gene batteries. This perhaps explains why a characteristic of the stem cell differentiation choice systems, in other words the simultaneous low level expression in the multipotent precursors of differentiation genes indicative of multiple possible fates48,49 (‘lineage priming’), is not seen in embryonic fate choices. That is, in embryonic body part development the spatial fate decision is made far up in the GRN hierarchy, and locked down, long before the differentiation gene battery is deployed. In contrast, in the production of functional immune cell types the last steps in the decision have to be deferred until the multipotential cells can be told which of its potentialities is more needed. Similar binary choice circuitry is also used in non-haematopoietic developmental contexts, but again at late stages in a given process where a terminal fate choice is to be made. For example, after mammalian somites have formed, they generate spatially confined subdomains, one of which is the dermomyotome. This consists of multipotent stem-like cells, where the choice to generate vascular muscle versus smooth muscle cell types is controlled by a modulated signal, mutual repression between the Pax3 and Foxc2 genes50. Another non-haematopoietic circuit that in essence is remarkably similar to the antagonistic haematopoietic repressor pair sub-circuits was discovered in C. elegans, also operating at the terminus of much prior development51. This circuit maintains the expression of distinct sets of differentiation genes expressed in left versus right taste neurons, but the duelling repressors expressed alternately in these two neurons are in this case microRNAs that directly target the mRNAs encoding the alternate differentiation drivers. All of these kinds of sub-circuits, operate to choose, and/or to maintain the choice, of one of an alternative pair of differentiation gene driver sets.
A priori, development of the body plan cannot be reduced to differentiated cell type specification, the last step in the process, nor to binary decisions between alternative fates. This is at root because development of the body plan requires a long sequence of multidimensional spatial decisions: during pattern formation spatial regulatory states must be installed progressively within multiple (>2) diverse boundaries, and also in certain anterior-posterior and dorsal-ventral positions with respect to the body plan. In each structure of the body regulatory states that include differentiation gene battery drivers are finally installed. Thus it is not in principle surprising that if the set of differentiation gene battery regulators is changed by experimental intervention, a different cell type can be made to appear. Many recent studies show that insertion of vectors expressing sets of transcription factors or even single transcription factors can result in the change of differentiated state from one haematopoietic cell type to another36; from fibroblast to neuron52, from exocrine to pancreatic β cell53, etc. These cell fate changes all occur near the far downstream periphery of GRN hierarchy, as symbolized in Fig. 1. Growing a new cell type requires activation of a new differentiation battery, whereas growing a new body part requires a prior process of spatial pattern formation driven by a deep GRN. More generally, although there are embryonic processes that look superficially like the binary choices just discussed, they are effected very differently. As an example, in the sea urchin embryo, endomesodermal precursor cells give rise both to mesoderm and to endoderm, fates driven by entirely distinct regulatory states. But a careful experimental analysis8 shows that there is no pluripotential ‘endomesodermal’ GRN, and instead a Delta/Notch signal activates a set of regulatory genes which constitute a mesoderm GRN, while in the same cells a Wnt/Tcf signal activates a different set of regulatory genes which constitute the endoderm GRN. The genes of the mesoderm GRN and of the endoderm GRN are expressed independently of one another, without any interactions. The cells of each regulatory state are then separated physically by a cell division, so that the Notch signal is received exclusively by one ring of cells, which becomes mesoderm, while the other cells express the endoderm GRN exclusively8. Nor are the exclusion functions (Table 1) that in given regulatory states act to repress genes key to alternative regulatory states ‘bipotential switches’. These sub-circuits are used to lock down regulatory choices already installed rather than to make choices. They may look superficially like the mutual repression sub-circuits that switch lineages bipotentially, but they are not.
Differentiation gene battery structure
Differentiation gene batteries account for functional cell type specificity, and a canonical network structure can be associated with them. This structure describes the topology of the regulatory relationships causing the protein coding differentiation genes of the battery to be expressed more or less coordinately. Differentiation gene batteries are per se shallow, relatively simply constructed types of sub-circuit, often wired in coherent feed forward format, as for example in sea urchin embryos1, pancreatic β-cells3, and macrophages54. As the immediately upstream GRNs are being uncovered, an additional characteristic of differentiation gene battery regulatory circuitry is often encountered: this is the occurrence of feedback between the drivers of the differentiation genes just upstream of the linkages to the effector genes, either auto- or cross-regulatory55,56, though this is not always seen26. The canonical form is that of Fig. 2a. Differentiation gene batteries consist of a sometimes very large number of effector genes, the relevant cis-regulatory modules of which (per battery) respond to members of a small set of transcription factors present as part of the terminal regulatory state. However, each such cis-regulatory module may in addition be serviced by some additional factors, which accounts for the fact that all the genes of the battery are not exactly expressed in lockstep3. For example, muscle protein genes are activated by two or three of the transcription factors orthologous to Srf, Mef2, and a myogenic bHLH factor in vertebrates57, plus, individually, other factors; whereas in C. elegans the differentiation genes of each class of neuron are identified by their response to a single key transcription factor, sometimes together with other factors55.
Figure 2. Structural characteristics of downstream effector gene cassettes and their control functions.

a, Typical differentiation gene battery, as discussed elsewhere3. Here each effector gene codes for a cell-type-specific protein required to generate the cell-specific output. These effector genes are all transcribed specifically in the given cell type in response to a small number of regulatory factors, which are themselves the output of the controlling specification GRN. Every effector gene of the battery is specifically controlled by these inputs. The immediate drivers of the battery shown cross-regulate (as is often the case). b, Structure that may be typical of morphogenetic effector gene cassettes. Here the output of the specification GRN is used to control transcription of only a minor fraction of key effector genes, and these in some way trigger or nucleate the process. But many of the proteins required for the function are widely expressed.
It is logically consistent that where there is direct repression of differentiation gene batteries by a proximal control circuit (‘anti-differentiation’) much the same architecture would be employed. In embryonic stem cells a hierarchical GRN that maintains the pluripotent state is headed by a recursive triple feedback system that links Nanog, Oct4 (also known as Pou5f1) and Sox2 genes58,59. Apparently directly downstream of this are linkages to many genes encoding transcriptional activators and repressors59,60, including a polycomb repressor that in turn targets regulatory genes associated with various differentiation states61. But also among the immediate targets of the triple feedback loop is the Rest gene, which encodes a factor that directly represses neurogenic differentiation genes62. This circuit is the mirror image of gene battery activation circuits.
Structure/function relations for GRNs controlling diverse kinds of biology
The downstream effector gene cassettes required for development include those executing morphogenetic cell biology functions, as well as differentiation gene batteries. A distinction is that by definition, differentiation genes are expressed cell type-specifically, whereas genes required for functions such as motility, ingression, invagination, cell division, convergent extension, tube formation, branching, shape remodelling, epithelial-mesenchyme transition, etc., may be deployed in many diverse cell types and many diverse contexts in development.
If we imagine a canonical differentiation gene battery to be structured as in Fig. 2a, how different will be the topology of a morphogenetic gene cassette? One possible clue comes from various studies on GRN linkages that execute transcriptional control of cell replication in developing systems. The spatial patterns of cell replication of course affect morphology, because the size and shape of given portions of a structure depend on the number of rounds of cell division mediated by the regulatory state in each developing region. In several cases the exact outputs of a developmental GRN that specifically control cell cycle activity have been determined. For example in developing pituitary, several linkages from the specification GRN directly control proliferation63: the Pitx1 gene provides inputs into the cyclin D1 gene; the Six1 gene acts to repress expression of a cell cycle arrest kinase; and Six1 plus other factors of the pituitary regulatory state activate c-myc (also known as Myc). In the developing zebrafish eye the GRN linkage to cell cycle control is regulation of cyclin D1 and c-myc (also known as myca/mycb) by the meis1 regulatory gene64. Thus, so to speak, these GRNs deploy the complex process of cell division by pressing a small number of regulatory ‘buttons’.
Perhaps only a subfraction of the effector genes in a morphogenetic gene cassette are transcriptionally regulated by direct inputs from the upstream GRN. This concept emerged from a study of the migration of heart precursor cells in developing Ciona65, one of the few system-level investigations we have into the transcriptional control of a morphogenetic function. A large number of cell biology genes participate in the processes of membrane protrusion and motility required for heart cell migration, but most of these genes are widely expressed. Migratory activity is specifically deployed by transcriptional activation of the rhoDF gene, which encodes a key required GTPase, and it is this gene which is directly controlled by the cis-regulatory outputs of the upstream GRN. The same principle is evident in a study of trichome formation in Drosophila66. Here again, an extensive patterning GRN lies upstream, and determines the location of the morphological features and its cellular progenitors. The remodelling of epidermal cell shape to produce trichomes (or alternately, smooth cuticle) is controlled by expression of the regulatory gene shavenbaby (also known as ovo), and some of its direct effector gene targets are known. But these are again only a fraction of the total genes whose products are required to build the trichome. If these examples are a guide, the wiring of differentiation gene batteries, in which every downstream gene is a specific target of the GRN (Fig. 2a), is distinct from the way morphogenetic gene cassettes may be wired (Fig. 2b). Many of the genes contributing to a morphogenetic cell biology process may be widely expressed and only a few key ‘button’ genes that functionally nucleate the whole process are transcriptionally controlled by GRN outputs, to deploy the process spatially. Were this a general result, it would promise the existence of simple regulatory levers by which morphogenetic cassettes could be re-deployed, either in evolution or in re-engineering projects, to which we return below.
A uniquely explanatory GRN analysis of innate immunity response mechanisms in dendritic cells, following stimulation of Toll-like receptors (TLRs)67, shows how a classic physiological response is programmed at the genomic level. Stimulation of TLRs 2, 3 and 4 with various agonists activates two partly overlapping response programs of effector gene expression, in other words an antiviral program and an inflammatory program. This study included all regulatory genes specifically involved in the process, and the architecture of the GRN was based on a comprehensive, quantitative perturbation analysis, using small hairpin RNAs (shRNAs) to block regulatory gene transcription, although no direct cis-regulatory validation of the GRN structure was reported. Several interesting differences and also similarities emerge in comparing the structure of this physiological response GRN to that of the developmental GRNs considered above. A salient similarity is in the structure of the effector gene sets. Like many differentiation genes, the TLR response effector gene sets are largely wired to their drivers in coherent feed forward loops. Another now familiar feature is the use of positive feedback that will lock down the regulatory state following a transient input, here between stat genes high up in the antiviral response GRN hierarchy. This GRN is of moderate depth: downstream of the stat genes are three other regulatory genes linked to the stat genes and to one another by cross-regulatory interactions, and downstream of these in turn are further regulatory genes, and then the effector genes. A further device in these GRNs that also is often used in development, is exclusion of the alternate regulatory state by specific cross-repression, once one of the pathways is active. The depth of the inflammatory hierarchy is only that of the feed forward circuitry. Physiological systems are homeostatic, and a special feature of this one is a self-cancelling repression circuit the sequence-specific basis of which is, however, yet unknown. Some years ago a prescient analysis predicted that in general, developmental GRNs which control progressive irreversible regulatory processes would have considerably greater depth than does reversible physiological response circuitry68, and this turns out to be exactly true.
One way of summarizing the result of a comparative meta-analysis of GRNs controlling diverse kinds of biological processes is to consider their similarities and differences in the same terms: they are similar in that all the GRNs considered here are modular constructs of a basic repertoire of sub-circuit topologies (Table 1); but they differ in their global hierarchical organization, which reasonably reflects the biological jobs they execute.
Insights into process from mathematical models of GRNs and sub-circuits
Space confines the following discussion to recently conceived models based ab initio on experimentally generated, largely validated network topologies. The major focus is on how, or whether, mathematical analyses of the models has succeeded in enriching our understanding of the biological functionalities of the observed circuitry.
Beginning with a known network topology, the common objective is to generate a dynamic mathematical model, either using continuous (ordinary differential equations or ODE) or Boolean approaches69. For large scale temporal models of embryonic spatial specification systems involving many genes and interactions, this often involves a great number of unmeasured parameters, and epistemological issues immediately arise. In many such works arbitrary parameter values are systematically explored until the expected results emerge, but this is inherently at least a partially circular logic, since it assumes a priori that the model is right. Of course where there are applicable experimental measurements of the output kinetics, the model is better constrained, but then the novelty of the biological insights that can be expected is limited because both the input relationships and the results are assumed. Drosophila gap gene expression in the syncytial embryo provides the best known large developmental data set thus far subjected to mathematical kinetic analysis70–72. Extensive genetic and cis-regulatory data partially specify the embryonic interaction networks of these genes73–75. Mathematical models were built assuming the network topologies proposed in prior work70,73, and fit to a very high quality set of quantitative kinetic measurements which capture the empirical dynamics of changing gap expression patterns in the pre-cellularization 13th–14th cleavage cycle71,72. There were two outcomes relevant to the structure/function relationships of this developmental GRN: First, a dynamic image of how the gap gene transcription factors operate emerged, illuminating what might be called the cell biology of the process (were there cells). Second, the analysis suggested several additions and corrections of unresolved details of gap gene interactions. But largely the outcome was just that if one does the math and the measurements, everything turns out to make sense.
An important area of developmental biology in which modelling has contributed novel mechanistic understanding is transcriptional response to signals. We cannot here deal with the many studies focused on dynamic spatial distributions of signal ligands per se. But mathematical analyses of models capturing transcriptional network circuitry downstream of intercellular signalling have illuminated developmental signal response in multiple ways. These models concern smaller and well constrained sub-circuits, rather than whole GRNs, and often either parameters can be reasonably approximated, or dimensionless approaches can be found. The signal-driven transcriptional patterning process by which the two dorsal respiratory appendages on the roof of the Drosophila egg are positioned affords an example76. An experimentally based network circuitry animated by spatially confined epidermal growth factor (EGF) and Dpp signalling was used to produce a dynamic mathematical model which satisfactorily interprets the changing pattern of expression of a key gene of the pro-appendage regulatory state in dorso-anterior follicle cells. The model thus explains how this system generates and positions the bilateral spots of gene expression where the appendages will form, which is not otherwise transparent. Furthermore, in consequence of a conflict between prediction and experimental observations, the analysis required a hitherto unsuspected positive feedback loop by which Dpp controls expression of its own receptor. A second example concerns transcriptional interpretation of graded hedgehog (Hh) signals in the developing neural tube, which results in a ventral to dorsal series of spatial regulatory state domains each of which gives rise to certain neuronal types34. When experimental measurements of signal intensity over time in the various transcriptional domains were analysed mathematically77,78, it emerged that the successive ventral to dorsal transcriptional domains are defined by the integrals over duration and intensity of Hh signalling, rather than simply on ‘morphogen concentration’, as always assumed previously. A third example79,80 relates to the Wnt signalling required in Xenopus embryos to activate key regulatory genes of the dorsal organizer. Experimental perturbations of this canonical developmental signalling system showed that this system responds to the ratio of the (signal) input at some given time, to its level when the signalling began (‘fold change’), and not to absolute signal level (the same phenomenon is often seen in other contexts, for example, sensory physiology). A predicted explanation in terms of network sub-circuit topology was then derived from a dynamic mathematical analysis of the incoherent feed forward sub-circuit80, which showed that this commonplace sub-circuit possesses the capacity to respond to fold change in input magnitude, rather than to absolute input magnitude.
As noted above, another general area in which modelling has illuminated process in respect to given sub-circuit topology is in binary cell fate choice, following a precursor phase in which both regulatory states are weakly expressed. Here the repeatedly observed sub-circuit structure features the opposition of two antagonistic repressors, each, if highly expressed, capable of shutting off the alternative regulatory state and generating its own, and each animated by inputs that reflect the external need for its pathway. A canonical approach to dynamic mathematical modelling of this type of sub-circuit has been repeatedly applied, based essentially on treating transcriptional activation abstractly as a catalytic Michaelis–Menton process, and repression in the same vein (for example, refs. 38, 81). The object is to demonstrate that these ‘duelling repressor’ sub-circuit topologies indeed encode regulatory systems that are capable of hysteretically moving from the precursor state to one or the other terminal regulatory states, depending on the inputs the system receives. But a problem with this approach is that as conventionally formulated, the bi-stable mathematical behaviour requires the completely ad hoc assumption of large exponential (Hill) coefficients in the repression functions (that is, coefficients >2, and often much larger values have to be assumed in order to generate the expected behaviour). Although Hill coefficients of these magnitudes physically imply cooperativity, or additional (unknown) reactions, they are customarily inserted in the computations despite lack of any direct biological evidence for cooperativity or other physical features that would justify them. Indeed, in one recent study of another very similarly wired haematopoietic choice system82, the erythroid/myeloid fate choice, it was pointed out that the specific, mutually repressive cis-regulatory interactions which were obtained are known not to be multimeric and cooperative, nor is there any other biochemical justification for high Hill coefficients. Instead an alternative regulatory architecture was considered, the dynamic mathematical analysis of which resulted in the prediction that the sub-circuit should include in addition to the antagonistic repressors another gene or genes operating according to specified network linkages. The latter work82, furthermore, used a now classic probabilistic thermodynamic treatment of cis-regulatory transcription factor binding83 that is directly based on transcription factor–DNA interaction physical chemistry. This same thermodynamic approach to modelling cis-regulatory transcription factor binding has been used for analysis of an entirely different type of sub-circuit operating at the initial developmental appearance of pluripotential haematopoietic stem cells28,47. This sub-circuit consists of three positively active genes. There are no cross-regulating repressors in this sub-circuit, and the three genes are linked by multiple positive auto- and cross-regulatory linkages. In life and in the model, extrinsic signals switch it irreversibly into an active state; otherwise, if one node is inhibited, it remains off47. Thus there are multiple different designs that confer signal-dependent bi-stability.
The thermodynamic binding approach83 was also used earlier for dynamic modelling of sea urchin embryo gene cascades84. The important insight emerged that in a cascade where a given gene activates a second downstream gene, significant expression of the second gene occurs long before the product of the first gene reaches steady state, and the whole dynamic system operates in a ‘forward drive mode’ relatively insensitive to levels of upstream activators. The kinetics of such embryonic regulatory interactions are not narrowly determinate, as emphasized by the kinetic ‘sloppiness’ of a process which operates successfully at different rates at different temperatures within and between similar species, and in which there is a significant range in the concentrations of many transcription factors embryo to embryo85.
For other situations in embryonic development where the object is to encompass a complex, large scale spatial specification system rather than to follow a given small domain or cell type through time, conventional, stand-alone dynamical analysis is the wrong tool for the job that really needs to be done. Returning to Table 1, for example, we see that there are several kinds of spatial specification sub-circuit, that in cellular early embryos produce novel spatial regulatory state patterns, for example X,1 − X spatial processing sub-circuits and AND spatial logic processors. These, and indeed many other embryonic specification processes that define multicellular territorial regulatory states, result in a progressive Boolean-like pattern of diverse regulatory states confronting one another sharply across territorial cellular boundaries. A model that would capture what the GRN really does must address this kind of outcome, capturing the encoded input information-processing behaviour at each cis-regulatory module of the GRN.
Current developmental GRNs mainly concern, on the one hand, far upstream hierarchical transactions that essentially execute regulatory state pattern formation, or on the other, far downstream differentiation gene batteries and their immediate governance. These will have to be much better linked, so that we have a continuous understanding of the control systems from the top of the hierarchy to all the effector genes of a developing system. This kind of global GRN will be much larger than anything we have at present. Other kinds of global GRNs are on the horizon as well, such as those that encompass all parts of a developing embryo through time. Experimentally validated GRNs that include complete large regulatory systems will present enormous computational challenges for modelling, presentation, logic analysis and modular abstraction.
Developmental GRNs and evolutionary mechanism
Because development of the body plan is caused by the operation of GRNs, evolutionary change in the body plan is change in GRN structure occurring over deep time86–88. Evolution and development emerge as twin outputs of the same mechanistic domain of regulatory system genomics. It is therefore to be expected that, at the level of GRN structure, each would illuminate the other, and so indeed they already do in several concrete ways. To start, it is obvious that if there is indeed a finite repertoire of network sub-circuits used to effect development, the evolution of development has to be considered as the process of assembly, reassembly, and redeployment of these sub-circuits. This general idea will become directly testable by widespread evolutionary comparisons, as the GRNs underlying the development of diverse animal forms become available. Structural comparison of GRNs between forms of known phylogenetic relation in turn reveals the modularity of GRN structure, by revealing sub-circuit boundaries, as when a sub-circuit is inserted wholesale into a new GRN context89. Furthermore, the sub-circuits of which GRNs are composed change during evolution at different rates, highlighting the linkages belonging to the most conserved sub-circuit in a GRN comparison. As discussed elsewhere, in general the oldest GRN features are certain differentiation gene batteries3,88, which are eumetazoan (cnidarian + bilaterian) in distribution. In contrast, the morphogenetic programs that pattern each form of body plan are by definition clade-specific88. Certain remarkably conserved regulatory sub-circuits that are located near the top of developmental GRN hierarchy may serve to lock down developmental process specific to given phyla or classes (GRN kernels)86–91. Thus GRNs are historically as well as structurally and functionally modular, in that they are a mosaic of sub-circuits of diverse antiquity and phylogenetic distribution. Systematic exploration of phylogenetically related GRNs at different distances is valuable not only to discover the evolutionary origins of each sub-circuit, but also to reveal which kinds of sub-circuits and linkages are inherently flexible and which not. This brings us to the most important point for the future. In order to probe control of spatial regulatory state, laboratory strategies will need to be designed for changing GRNs by insertion of network regulatory apparatus into developing systems. But this is the same kind of change that happened in evolution, and the results will be mutually informative. Thus a practical convergence is on the horizon. Re-engineering spatial developmental processes, and recreating evolutionary processes, while different in motivation, will both depend on fundamental understanding and experimental manipulation of the structure/function relations of developmental GRNs.
The processes we have been discussing, development and evolution of the body plan, and execution of physiological responses, devolve causally from the regulatory genome. We need to understand GRNs because they encompass the primary output of the regulatory genome, itself the fundamental and unique outcome of more than 600 million years of animal evolution88.
Acknowledgements
I am grateful for the reviews of the manuscript by E. V. Rothenberg and I. S. Peter. This work was supported by NIH grants HD-37105 and GM-61005 and by the Lucille P. Markey Charitable Trust.
Footnotes
The author declares no competing financial interests.
References
- 1. Oliveri P, Tu Q, Davidson EH. Global regulatory logic for specification of an embryonic cell lineage. Proc. Natl Acad. Sci. USA. 2008;105:5955–5962. doi: 10.1073/pnas.0711220105. This paper provides proof of principle that if a developmental GRN is essentially complete, then it provides causal explanations for the biological functions of the process it controls.
- 2. Peter IS, Davidson EH. Modularity and design principles in the sea urchin embryo gene regulatory network. FEBS Lett. 2009;583:3948–3958. doi: 10.1016/j.febslet.2009.11.060. This paper presents the latest comprehensive review of the sea urchin endomesoderm GRN, so far the most extensively validated large scale embryonic GRN, with special emphasis on the topologies of its spatial control sub-circuits.
- 3.Davidson EH. The Regulatory Genome. Gene Regulatory Networks in Development and Evolution. Elsevier: Academic Press; 2006. [Google Scholar]
- 4.Alon U. Network motifs: theory and experimental approaches. Nature Rev. Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 5.Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc. Natl Acad. Sci. USA. 2003;100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davidson EH. Network design principles from the sea urchin embryo. Curr. Opin. Genet. Dev. 2009;19:535–540. doi: 10.1016/j.gde.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma W, Trusina A, El-Samad H, Lim WA, Tang C. Defining network topologies that can achieve biochemical adaptation. Cell. 2009;138:760–773. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peter IS, Davidson EH. The endodermgene regulatory network in sea urchin embryos up to mid-blastula stage. Dev. Biol. 2010;340:188–199. doi: 10.1016/j.ydbio.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oliveri P, Davidson EH. Built to run, not fail. Science. 2007;315:1510–1511. doi: 10.1126/science.1140979. [DOI] [PubMed] [Google Scholar]
- 10.Koide T, Hayata T, Cho KWY. Xenopus as a model system to study transcriptional regulatory networks. Proc. Natl Acad. Sci. USA. 2005;102:4943–4948. doi: 10.1073/pnas.0408125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maduro MF. Structure and evolution of the C. elegans embryonic endomesoderm network. Biochim. Biophys. Acta. 2009;1789:250–260. doi: 10.1016/j.bbagrm.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan T-M, et al. Developmental gene regulatory networks in the zebrafish embryo. Biochim. Biophys. Acta. 2009;1789:279–298. doi: 10.1016/j.bbagrm.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Morley RH, et al. A gene regulatory network directed by zebrafish No tail accounts for its roles in mesoderm formation. Proc. Natl Acad. Sci. USA. 2009;106:3829–3834. doi: 10.1073/pnas.0808382106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stathopoulos A, Levine M. Genomic regulatory networks and animal development. Dev. Cell. 2005;9:449–462. doi: 10.1016/j.devcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 15. Hong J-W, Hendrix DA, Papatsenko D, Levine MS. How the Dorsal gradient works: insights from postgenome technologies. Proc. Natl Acad. Sci. USA. 2008;105:20072–20076. doi: 10.1073/pnas.0806476105. This review summarizes work regulatory control of Dorsal target genes expressed spatially along the dorsal/ventral axis of the syncytial Drosophila embryo.
- 16. Liberman LM, Stathopoulos A. Design flexibility in cis-regulatory control of gene expression: synthetic and comparative evidence. Dev. Biol. 2009;327:578–589. doi: 10.1016/j.ydbio.2008.12.020. This paper presents a novel experimental evidence of cis-regulatory design features in the syncytial dorsal-ventral Drosophila specification system.
- 17.Levine M, Davidson EH. Gene regulatory networks for development. Proc. Natl Acad. Sci. USA. 2005;102:4936–4942. doi: 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ochoa-Espinosa A, Yu D, Tsirigos A, Struffi P, Small S. Anterior-posterior positional information in the absence of a strong Bicoid gradient. Proc. Natl Acad. Sci. USA. 2009;106:3823–3828. doi: 10.1073/pnas.0807878105. This paper provides experimental evidence that the anterior/posterior specification system of the Drosophila embryo is controlled by a network of gene interactions rather than only by quantitative positional values of Bicoid.
- 19.Liberman LM, Teeves GT, Stathopoulos A. Quantitative imaging of the Dorsal nuclear gradient reveals limitations to threshold-dependent patterning in Drosophila. Proc. Natl Acad. Sci. USA. 2009;106:22317–22322. doi: 10.1073/pnas.0906227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang AM, Rusch J, Levine M. An anteroposterior Dorsal gradient in the Drosophila embryo. Genes Dev. 1997;11:1963–1973. doi: 10.1101/gad.11.15.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saka Y, Smith JC. A mechanism for sharp transition of morphogen gradient interpretation in Xenopus. BMC Dev. Biol. 2007;7:47–55. doi: 10.1186/1471-213X-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davidson EH. Genomic Regulatory Systems: Development and Evolution. Elsevier: Academic Press; 2001. [Google Scholar]
- 23.Su Y-H, et al. A perturbation model of the gene regulatory network for oral and aboral ectoderm specification in the sea urchin embryo. Dev. Biol. 2009;329:410–421. doi: 10.1016/j.ydbio.2009.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikitina N, Sauka-Spengler T, Bronner-Fraser M. Dissecting early regulatory relationships in the lamprey neural crest gene network. Proc. Natl Acad. Sci. USA. 2008;105:20083–20088. doi: 10.1073/pnas.0806009105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woodland HR, Zorn AM. The core endodermal gene network of vertebrates: combining developmental precision with evolutionary flexibility. Bioessays. 2008;30:757–765. doi: 10.1002/bies.20785. [DOI] [PubMed] [Google Scholar]
- 26.Cvekl A, Duncan MK. Genetic and epigenetic mechanisms of gene regulation during lens development. Prog. Retin. Eye Res. 2007;26:555–597. doi: 10.1016/j.preteyeres.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar JP. The molecular circuitry governing retinal determination. Biochim. Biophys. Acta. 2009;1789:306–314. doi: 10.1016/j.bbagrm.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pimanda JE, et al. Gata2, Fli1, and Scl form a recursively wired gene-regulatory circuit during early hematopoietic development. Proc. Natl Acad. Sci. USA. 2007;104:17692–17697. doi: 10.1073/pnas.0707045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith PA, Mango SE. Role of T-box gene tbx-2 for anterior foregut muscle development in C. elegans. Dev. Biol. 2007;302:25–39. doi: 10.1016/j.ydbio.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cripps RM, Olson EN. Control of cardiac development by an evolutionarily conserved transcription network. Dev. Biol. 2002;246:14–28. doi: 10.1006/dbio.2002.0666. [DOI] [PubMed] [Google Scholar]
- 31.Reim I, Mohler JP, Frasch M. Tbx20-related genes, mid and H15 are required for tinman expression, proper patterning, and normal differentiation of cardioblasts in Drosophila. Mech. Dev. 2005;122:1056–1069. doi: 10.1016/j.mod.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Albert R, Othmer HG. The topology of the regulatory interactions predicts the expression pattern of the segment polarity genes in Drosophila melanogaster. J. Theor. Biol. 2003;223:1–18. doi: 10.1016/s0022-5193(03)00035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishi Y, Ji H, Wong WH, McMahon AP, Vokes SA. Modeling the spatiotemporal network that drives patterning in the vertebrate central nervous system. Biochim. Biophys. Acta. 2009;1789:299–305. doi: 10.1016/j.bbagrm.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Vokes SA, Ji H, Wong WH, McMahon AP. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 2008;22:2651–2663. doi: 10.1101/gad.1693008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ririe TO, Fernandes JS, Sternberg PW. The Caenorhabditis elegans vulva: A post-embryonic gene regulatory network controlling organogenesis. Proc. Natl Acad. Sci. USA. 2008;105:20095–20099. doi: 10.1073/pnas.0806377105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Graf T, Enver T. Forcing cells to change lineages. Nature. 2009;462:587–594. doi: 10.1038/nature08533. This review comprehensively traverses the process of terminal lineage fate choice in pluripotential hematopoietic systems.
- 37.Swiers G, Patient R, Loose M. Genetic regulatory networks programming hematopoietic stem cells and erythroid lineage specification. Dev. Biol. 2006;294:525–540. doi: 10.1016/j.ydbio.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 38. Laslo P, et al. Multilineage transcription priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. This paper exemplifies a commonly used mathematical approach invoking bi-stable state kinetics to explain lineage choice.
- 39.Smith J, Davidson EH. Gene regulatory network subcircuit controlling a dynamic spatial pattern of signaling in the sea urchin embryo. Proc. Natl Acad. Sci. USA. 2008;105:20089–20094. doi: 10.1073/pnas.0806442105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang P, et al. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc. Natl Acad. Sci. USA. 1999;96:8705–8710. doi: 10.1073/pnas.96.15.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang S, Guo Y-P, May G, Enver T. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev. Biol. 2007;305:695–713. doi: 10.1016/j.ydbio.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 42.Stopka T, Amanatullah DF, Papetti M, Skoultchi AI. PU.1 inhibits the erythroid program by binding to GATA-1 on DNA and creating a repressive chromatin structure. EMBO J. 2005;24:3712–3723. doi: 10.1038/sj.emboj.7600834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Starck J, et al. Functional cross-antagonism between transcription factors FLI-1 and EKLF. Mol. Cell. Biol. 2003;23:1390–1402. doi: 10.1128/MCB.23.4.1390-1402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rothenberg EV. Decision by committee: new light on the CD4/CD8-lineage choice. Immunol. Cell Biol. 2009;87:109–112. doi: 10.1038/icb.2008.100. [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Bosselut R. CD4–CD8 lineage differentiation: Thpok-ing into the nucleus. J. Immunol. 2009;183:2903–2910. doi: 10.4049/jimmunol.0901041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Setoguchi R, et al. Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science. 2008;319:822–825. doi: 10.1126/science.1151844. [DOI] [PubMed] [Google Scholar]
- 47.Narula J, Smith AM, Gottgens B, Igoshin OA. Modeling reveals bistability and low-pass filtering in the network module determining blood stem cell fate. PLoS Comp. Biol. 2010;6:e1000771. doi: 10.1371/journal.pcbi.1000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu M, et al. Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 1997;11:774–785. doi: 10.1101/gad.11.6.774. [DOI] [PubMed] [Google Scholar]
- 49.Miyamoto T, et al. Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev. Cell. 2002;3:137–147. doi: 10.1016/s1534-5807(02)00201-0. [DOI] [PubMed] [Google Scholar]
- 50.Lagha M, et al. Pax3:Foxc2 reciprocal repression in the somite modulates muscular versus vascular cell fate choice in multipotent progenitors. Dev. Cell. 2009;17:892–899. doi: 10.1016/j.devcel.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 51.Johnson RJ, Jr, Chang S, Etchberger JF, Ortiz CO, Hobert O. MicroRNAs acting in a double-negative feedback loop to control a neuronal cell fate decision. Proc. Natl Acad. Sci. USA. 2005;102:12449–12454. doi: 10.1073/pnas.0505530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vierbuchen T, et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilchrist M, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 55.Hobert O. Regulatory logic of neuronal diversity: Terminal selector genes and selector motifs. Proc. Natl Acad. Sci. USA. 2008;105:20067–20071. doi: 10.1073/pnas.0806070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bröhl D, et al. A transcriptional network coordinately determines transmitter and peptidergic fate in the dorsal spinal chord. Dev. Biol. 2008;322:381–393. doi: 10.1016/j.ydbio.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Yun K, Wold B. Skeletal muscle determination and differentiation: story of a core regulatory network and its context. Curr. Opin. Cell Biol. 1996;8:877–889. doi: 10.1016/s0955-0674(96)80091-3. [DOI] [PubMed] [Google Scholar]
- 58.Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17:42–49. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- 59.Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Q, Chipperfield H, Melton DA, Wong WH. A gene regulatory network in mouse embryonic stem cells. Proc. Natl Acad. Sci. USA. 2007;104:16438–16443. doi: 10.1073/pnas.0701014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 62.Mortazavi A, Chen Leeper Thompson E, Garcia ST, Myers RM, Wold B. Comparative genomics modeling of the NRSF/REST repressor network: from single conserved sites to genome-wide repertoire. Genome Res. 2006;16:1208–1221. doi: 10.1101/gr.4997306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu X, Rosenfeld MG. Transcriptional control of precursor proliferation in the early phases of pituitary development. Curr. Opin. Genet. Dev. 2004;14:567–574. doi: 10.1016/j.gde.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 64.Bessa J, et al. meis1 regulates cyclin D1 and c-myc expression, and controls the proliferation of the multipotent cells in the early developing zebrafish eye. Development. 2008;135:799–803. doi: 10.1242/dev.011932. [DOI] [PubMed] [Google Scholar]
- 65. Christiaen L, et al. The transcription/migration interface in heart precursors of Ciona intestinalis. Science. 2008;320:1349–1352. doi: 10.1126/science.1158170. This paper presents direct evidence of the regulatory structure of a morphogenetic gene cassette, showing that only certain key genes are controlled by the upstream GRN while a majority are expressed anyway.
- 66.Chanut-Delalande H, Fernandes I, Roch F, Payre F, Plaza S. Shavenbaby couplespatterning to epidermal cell shape control. PLoS Biol. 2006;4:1549–1561. doi: 10.1371/journal.pbio.0040290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Amit I, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. This paper presents the most complete analysis yet available of structure and function in a physiological GRN.
- 68.Rosenfeld N, Alon U. Response delays and the structure of transcription networks. J. Mol. Biol. 2003;329:645–654. doi: 10.1016/s0022-2836(03)00506-0. [DOI] [PubMed] [Google Scholar]
- 69.Bolouri H. Computational Modeling of Gene Regulatory Networks – A Primer. Imperial College Press; 2008. [Google Scholar]
- 70.Sánchez L, Thieffry D. A logical analysis of the gap gene system. J. Theor. Biol. 2001;211:115–141. doi: 10.1006/jtbi.2001.2335. [DOI] [PubMed] [Google Scholar]
- 71.Jaeger J, et al. Dynamical analysis of regulatory interactions in the gap gene system of Drosophila melanogaster. Genetics. 2004;167:1721–1737. doi: 10.1534/genetics.104.027334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Perkins TJ, Jaeger J, Reintz J, Glass L. Reverse engineering the gap gene network of Drosophila melanogaster. PLoS Comp. Biol. 2006;2:e051. doi: 10.1371/journal.pcbi.0020051. This paper provides a comprehensive computational treatment of the Drosophila gap gene network using estimates of numerous constants obtained by high resolution imaging.
- 73.Rivera-Pomar R, Jaeckle H. From gradients to stripes in Drosophila mebryogenesis: filling in the gaps. Trends Genet. 1996;12:478–483. doi: 10.1016/0168-9525(96)10044-5. [DOI] [PubMed] [Google Scholar]
- 74.Kraut R, Levine M. Mutually repressive interactions between the gap genes giant and Krüpple define middle body regions of the Drosophila embryo. Development. 1991;111:611–621. doi: 10.1242/dev.111.2.611. [DOI] [PubMed] [Google Scholar]
- 75.Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U. Predicting expression patterns fromregulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- 76.Lembong J, Yakoby N, Shvartsman SY. Pattern formation by dynamically interacting network motifs. Proc. Natl Acad. Sci. USA. 2009;106:3213–3218. doi: 10.1073/pnas.0810728106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dessaud E, et al. Dynamic assignment and maintenance of positional identity in the ventral neural tube by the morphogen Sonic hedgehog. PLoS Biol. 2010;8:e1000382. doi: 10.1371/journal.pbio.1000382. This paper provides a new insight into how positional values of Hedgehog ligand are used to set transcriptional thresholds.
- 78.Ribes V, Briscoe J. Establishing and interpreting graded Sonic Hedgehog signaling during vertebrate neural tube patterning: the role of negative feedback. Cold Spring Harb. Perspect. Biol. 2009;1:a002014. doi: 10.1101/cshperspect.a002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Goentoro L, Shoval O, Kirschner MW, Alon U. The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol. Cell. 2009;36:894–899. doi: 10.1016/j.molcel.2009.11.018. This analysis shows how a common GRN sub-circuit can operate to interpret relative changes in signal strength.
- 80.Goentoro L, Kirschner MW. Evidence that fold-change, and not absolute level, of β-catenin dictates Wnt signaling. Mol. Cell. 2009;36:872–884. doi: 10.1016/j.molcel.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spooner CJ, et al. A recurrent network involving the transcription factors PU.1 and Gfi1 orchestrates innate and adaptive immune cell fates. Immunity. 2009;31:576–586. doi: 10.1016/j.immuni.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chickarmane V, Enver T, Peterson C. Computational modeling of the hematopoietic erythroid-myeloid switch reveals insights into cooperativity, priming, and irreversibility. PLoS Comp. Biol. 2009;5:e1000268. doi: 10.1371/journal.pcbi.1000268. This paper presents an alternative computational treatment of lineage choice in a haematopoietic system.
- 83.Shea MA, Ackers GK. The OR control system of bacteriophage lambda: A physical-chemical model for gene regulation. J. Mol. Biol. 1985;181:211–230. doi: 10.1016/0022-2836(85)90086-5. [DOI] [PubMed] [Google Scholar]
- 84. Bolouri H, Davidson EH. Transcriptional regulatory cascades in development: Initial rates, not steady state, determine network kinetics. Proc. Natl Acad. Sci. USA. 2003;100:9371–9376. doi: 10.1073/pnas.1533293100. This paper models sea urchin regulatory cascade kinetics and demonstrates using measured constants that genes are successively activated long before any of the transcriptional functions attain steady state.
- 85.Materna SC, Nam J, Davidson EH. High accuracy, high-resolution prevalence measurement for the majority of locally expressed regulatory genes in early sea urchin development. Gene Expr. Patterns. 2010;10:177–184. doi: 10.1016/j.gep.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Davidson EH, Erwin DH. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. This paper introduced the theory that highly conserved GRN sub-circuits account for the phylogenetic distribution of major characters of the animal body plan.
- 87.Erwin DH, Davidson EH. The evolution of hierarchical gene regulatory networks. Nature Rev. Genet. 2009;10:141–148. doi: 10.1038/nrg2499. [DOI] [PubMed] [Google Scholar]
- 88.Davidson EH, Erwin DH. An integrated view of Precambrian eumetazoan evolution. Cold Spring Harb. Symp. Quant. Biol. 2009;74:65–80. doi: 10.1101/sqb.2009.74.042. [DOI] [PubMed] [Google Scholar]
- 89.Gao F, Davidson EH. Transfer of a large gene regulatory apparatus to a new developmental address in echinoid evolution. Proc. Natl Acad. Sci. USA. 2008;105:6091–6096. doi: 10.1073/pnas.0801201105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hinman VF, Davidson EH. Evolutionary plasticity of developmental gene regulatory network architecture. Proc. Natl Acad. Sci. USA. 2007;104:19404–19409. doi: 10.1073/pnas.0709994104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hinman VF, Yankura KA, McCauley BS. Evolution of gene regulatory network architectures: Examples of subcircuit conservation and plasticity between classes of echinoderms. Biochim. Biophys. Acta. 2009;1789:326–332. doi: 10.1016/j.bbagrm.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 92.Bolouri H, Davidson EH. The gene regulatory network basis of the “community effect,” and analysis of a sea urchin embryo example. Dev. Biol. 2010;340:170–178. doi: 10.1016/j.ydbio.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]