




Summary
CRISPR pools are being widely employed to identify gene functions. However, current technology, which utilizes DNA as barcodes, permits limited phenotyping and bulk-cell resolution. To enable novel screening capabilities, we developed a barcoding system operating at the protein level. We synthesized modules encoding triplet combinations of linear epitopes to generate >100 unique protein barcodes (Pro-Codes). ProCode-expressing vectors were introduced into cells and analyzed by CyTOF mass-cytometry. Using just 14 antibodies, we detected 364 Pro-Code populations; establishing the largest set of protein-based reporters. By pairing each Pro-Code with a different CRISPR, we simultaneously analyzed multiple phenotypic markers, including phospho-signaling, on dozens of knockouts. Pro-Code/CRISPR screens found two interferon-stimulated genes, the immunoproteasome component Psmb8 and a chaperone Rtp4, are important for antigen-dependent immune editing of cancer cells, and identified Socs1 as a negative regulator of Pd-l1. The Pro-Code technology enables simultaneous high-dimensional protein-level phenotyping of 100s of genes with single cell resolution.
In Brief
Protein-level genetic barcodes enable single-cell high-dimensional phenotyping by mass cytometry in CRISPR screens
Graphical Abstract
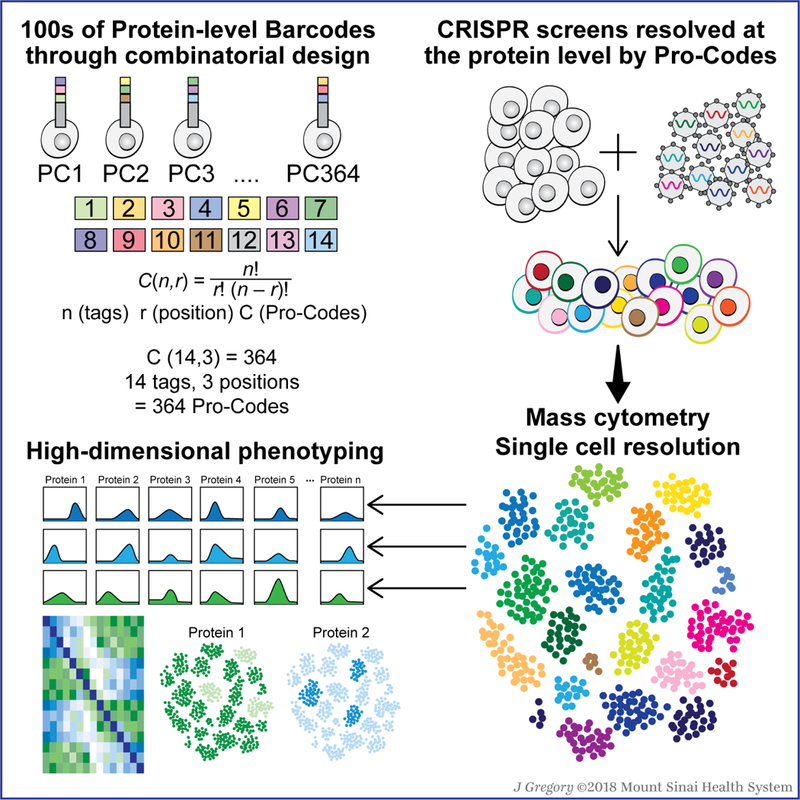
Introduction
There are more than 20,000 protein-coding genes in the human genome, as well as 100s of non-coding RNA genes, including microRNAs. Though there has been progress in assigning functions to many genes, we still do not know all the functions of each gene, or the role of many genes in driving or affecting disease. Determining the functions of every gene, in different normal and disease processes, is one of the major goals of the post-genome era.
The technology exists to knockout (KO), knockdown (KD), or overexpress (OE) any gene using vectors encoding a CRISPR guide RNA (gRNA) or shRNA. However, KO, KD, or OE of every gene in a genome in distinct experimental systems is cumbersome, costly, and very time consuming. For in vivo studies, it is even more challenging, and not practically feasible. This has led to the increasing use of pooled genetic screens aimed at determining the functions of 100s of genes simultaneously in a single experimental system. Pooled screens have been made possible by using DNA to barcode vectors. Unique nucleotide sequences can be incorporated in to a vector, or alternatively, when the vector encodes an shRNA or CRISPR gRNA, the shRNA or gRNA sequence becomes the barcode (Bassik et al., 2009; Shalem et al., 2015). Cells can be transduced with 100s of vectors simultaneously, and the frequency of cells carrying each vector can be determined by deep-sequencing (Mullokandov et al., 2012). The function of a particular gene is inferred by applying a selective pressure, such as time or a drug, and measuring changes in the frequency of each barcode associated with a particular shRNA or gRNA.
DNA barcoding has major limitations. One of the most significant is that the read-out is performed on bulk cells, which means single cells cannot be readily analyzed. This is a problem for many reasons, but one is that KO, KD, and OE does not occur in 100% of cells, and thus analyzing in bulk includes a mix of cells with and without the genetic perturbation. Another limitation is that DNA barcoding does not enable cells to be directly phenotyped. Instead, the phenotype associated with each gene perturbation is inferred from changes in barcode frequency. This has limited pooled screens largely to vetting genes for their potential impact on cell fitness, and inferring a change in shRNA/gRNA frequency is due to KD/KO influencing proliferation or survival (Shalem et al., 2015). More informative phenotypes, such as upregulation or downregulation of specific proteins, cannot be easily assessed in screens using DNA barcodes. Recently, CRISPR screens have been coupled with single cell RNA sequencing (scRNA-seq), and vector-encoded RNA used as a barcode (Adamson et al., 2016; Datlinger et al., 2017; Dixit et al., 2016; Jaitin et al., 2016). This enabled more high content and high-resolution screens. However, the cell throughput is relatively restrained, and important protein-level phenotypic information, such as signaling alterations, cannot be measured.
Here we show that combinatorial arrangements of linear epitopes can be used to generate a protein barcoding system (Pro-Codes), which is capable of overcoming many limitations of current pooled screening technology. We synthesized sequences encoding 3 combinations of 14 different linear epitopes to create 364 Pro-Codes. Pro-Code-expressing vectors were introduced into cells, and we could simultaneously detect all 364 Pro-Code-expressing cell populations. By pairing each Pro-Code with a different CRISPR gRNA, we were able to analyze multiple proteins on dozens of knockouts with single cell resolution. We used Pro-Code/CRISPR vectors to screen for genes that influence breast cancer sensitivity and resistance to antigen-specific T cell killing, and found evidence that two interferon-gamma (IFNγ) stimulated genes, the immunoproteasome component Psmb8 and a poorly characterized chaperone Rtp4, are important for antigen-dependent immune editing. Within the same screen, we also found that Socs1 is a negative regulator of the immune checkpoint Pd-l1. This work establishes a new barcoding system that enables simultaneous high dimensional phenotypic analysis of 100s of genes, at single cell resolution, with broad applications for helping to advance gene annotation.
Results
Pro-Codes enable highly multiplexed cell barcoding at the protein level
We sought to generate a vector barcoding system that operates at the protein level, as this would allow us to multiplex many gene delivery vectors together, and detect them in cells using high-throughput, single cell resolution technologies, such as flow and mass cytometry, and enable complex phenotyping. Proteins such as GFP and RFP can be used as vector reporters, but each fluorescent protein requires its own detection channel, which limits the number of unique fluorescent reporters that can be used together, generally to 3 or 4, since fluorescent proteins have broad emission spectrums that can overlap. To solve this problem, we hypothesized that combinations of a limited number of antibody-detectable epitopes (n) could be arranged together in specific multiples (r) to form a higher order set of barcodes (C) (Figure 1A). Using this strategy, as few as 10 epitopes can be arranged in sets of 3 to create 120 unique combinations, and with just 20 epitopes and 7 positions, 77,520 combinations can be generated.
Figure 1. Single cell analysis of 120 Pro-Code expressing populations.
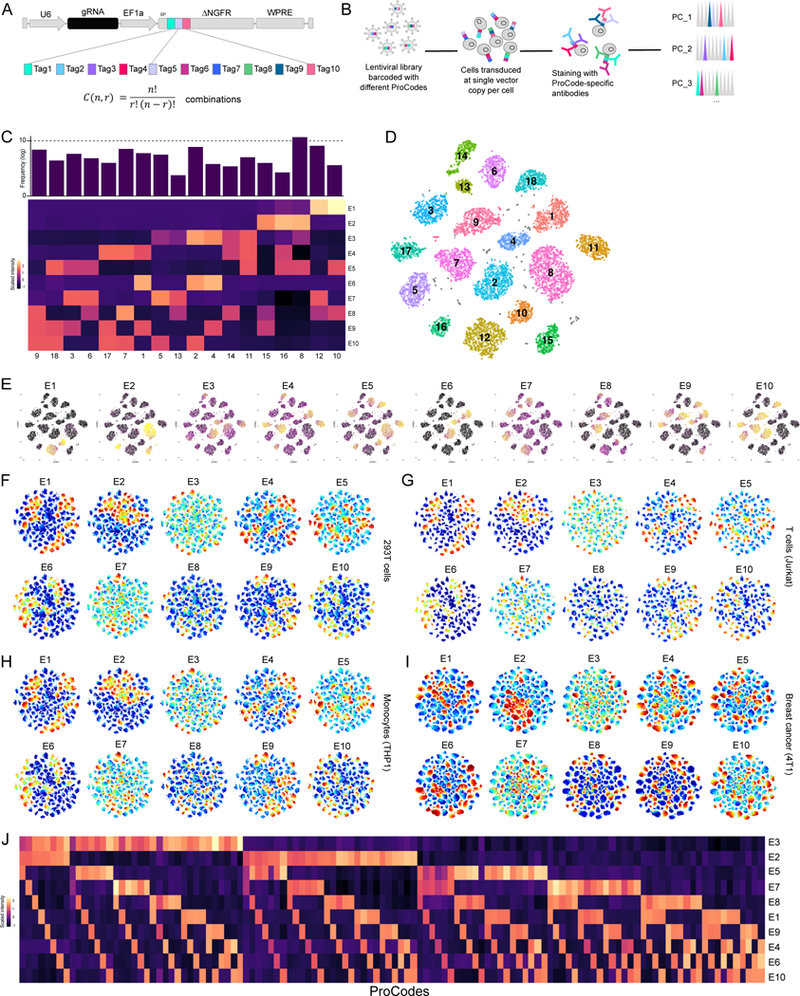
(A) Schematic of the Pro-Code vectors. (n) linear epitopes, (r) positions, (C) Pro-Codes. (B) Schematic of transduction, staining, and analysis. (C) 293T cells were transduced with 18 different Pro-Code vectors, stained for each epitope, and analyzed by CyTOF. Heatmap of relative expression of each epitope (E1-E10). (D) viSNE clustering of data in (C). (E) viSNE plots showing expression of each epitope from (C). Expression is scaled from high to low (yellow to dark purple). (F) 293T, (G) Jurkat, (H) THP1 and (I) 4T1 were transduced with a pool of 120 Pro-Codes vectors, and analyzed by CyTOF. Shown is the viSNE clustering with expression of each epitope (E1–10) colored from high to low (red to blue). (J) Heatmap showing epitope (E) expression for each of the 120 Pro-Code populations in 293T. All data is representative of 3 independent experiments.
We reasoned linear epitopes would be needed to assemble the barcodes because they can be encoded by a short sequence (18–42 nucleotides). We selected 10 linear epitopes for which there are antibodies for detection. Amongst these were epitopes commonly used as protein tags, such as HA and FLAG (Table S1). We synthesized the DNA sequence encoding each epitope, and assembled them in every possible combination of 3, for a total of 120 different 3-epitope combinations. We fused each epitope combination to dNGFR, a truncated receptor without an intracellular domain, which is commonly used as a reporter protein (Mullokandov et al., 2012). This served as a scaffold to facilitate epitope transport to the cell surface (Figure 1A). Each of the 120 3-epitope/dNGFR combinations (herein referred to as Pro-Codes) were cloned into lentiviral vectors (LV) downstream of the EF1a promoter. Vector plasmids were pooled in equimolar ratio, and used to make a pool of LV encoding the Pro-Codes.
We transduced 293T kidney cells with a pool of 18 LVs each encoding a different 3-epitope Pro-Code. The cells were transduced at a low multiplicity of infection (MOI) so each cell was only transduced with a single Pro-Code vector. The cells were then harvested and stained with antibodies for NGFR and all 10 of the linear epitopes. Each of the antibodies were conjugated with a different metal, and samples were analyzed on a CyTOF mass cytometer (Figure 1B). We used mass cytometry because CyTOF permits detection of over 45 different metal-conjugated antibodies (Bendall et al., 2011), and would thus enable detection of the Pro-Code epitopes along with more than 35 phenotypic markers. All 10 epitope tags were detected with a clear signal over background, and all of the epitope-positive cells were positive for NGFR (Figure S1A).
To determine if we could resolve cells expressing specific Pro-Codes, we analyzed NGFR+ cells using a debarcoder algorithm (Fread et al., 2017). Eighteen distinct cell populations were detected (Figure 1C and S1B), with each population corresponding to a unique Pro-Code (i.e. positive for precisely 3 of the 10 epitopes). For example, one population of cells was positive for the E3, E4, and E5 epitopes, and negative for all other epitopes, indicating the cells expressed the E3-E4-E5 Pro-Code (Figure S1C). We clustered NGFR+ cells based on their epitope tag expression. Once again 18 distinct populations were identified with each cluster positive for only 3 epitopes, and thus corresponding precisely to a specific Pro-Code (Figure 1D, E). To determine if we could increase the number of epitopes per Pro-Code, we generated 14 Pro-Codes with 4 epitopes per Pro-Code. We cloned each one in to an LV, transduced 293T with the pool, and analyzed by CyTOF. We detected all 10 epitopes, and cells were positive for 4 epitopes. This enabled us to identify all 14 4-epitope Pro-Code populations (Figure S1D).
Next we pooled 120 different 3-epitope Pro-Code plasmids together in a roughly equimolar ratio, and made a library of LV. We transduced 293T, as well as monocytic cells (THP1), leukemic T cells (Jurkat), and mammary carcinoma cells (4T1) with the 120 vector library. After 1 week, cells were stained with the 10 metal-conjugated antibodies, and analyzed by CyTOF. Unsupervised clustering resolved 120 distinct populations (Figure 1F-I), with each population corresponding to one Pro-Code (Figure 1J and S2A-C). The frequency of each population ranged from 0.1% to 3%, with the majority of Pro-Code populations (65%) being between 0.4–1.5% (Figure S2D), which is close to the expected frequency of 0.83% if each of the 120 Pro-Codes was in equimolar concentration.
Using an expanded set of 14 epitopes, we generated 364 3-epitope Pro-Code vectors, and introduced them in to 293T. The cells were stained for NGFR and all 14 epitopes, analyzed by CyTOF, and all 364 Pro-Code expressing populations were readily identified and clustered (Figure S3). Thus, with only 14 antibodies (i.e. 14 detection channels), we could detect 364 different vector expressing cell populations. These results demonstrate that combinations of linear epitopes can be used to generate protein barcodes detectable at the protein level and at single-cell resolution.
Pro-Codes can be used in vivo to track cancer cell clonality
One important application of vector barcoding is in cell clone and lineage tracing (Lu et al., 2011). Fluorescent proteins provide a powerful way to do this, but the number of populations that can be tracked is quite limited. DNA barcodes can tag an almost infinite number of cells, but only provide bulk resolution. The Pro-Codes could potentially be used for clone tracking, but an important requirement is in vivo use. To address this, we transduced 4T1 mammary carcinoma cells with a pool of 120 Pro-Code vectors at low MOI. Cells were sorted based on NGFR, as dNGFR serves not only as a Pro-Code scaffold, but also as a selectable marker of transduced cells. The transduced cells were injected in to the right and left mammary gland of wildtype (WT) mice (n=5 mice, 2 tumors per mouse) (Figure 2A). Since cells expressing non-self-proteins can be subject to immune clearance in immunocompetent animals, we also injected Rag1-/” immunodeficient mice for comparison (n=6 mice, 2 tumors per mouse).
Figure 2. Analysis of Pro-Code labeled breast tumors.
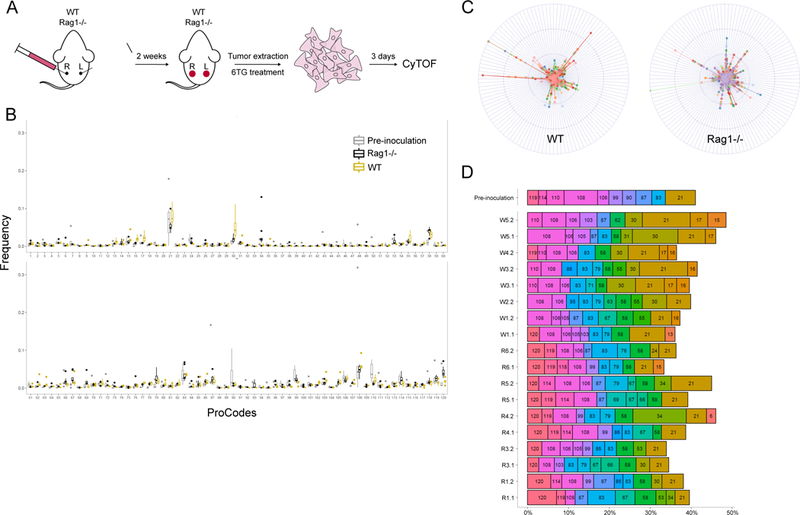
(A) Schematic of the in vivo 4T1 tumor studies. (B) Frequency of each Pro-Code population in tumors from wild-type and Rag1−/− mice. Shown is the median ± interquartile range (8–10 tumors/mouse group). Also included is the frequency of each Pro-Code in the 4T1 cells prior to inoculation (Pre-inoculation). (C) Distribution of the Pro-Code populations among each tumor. Data is presented in radar plots. The distance from the center represents the frequency of a Pro-Code population (each color represents a tumor, each quadrant corresponds to cells expressing a different Pro-Code). (D) Frequency of the 10 most abundant Pro-Code populations in each individual tumor. On the Y axis are individual tumors from WT (W) or Rag1-/” (R) mice. Numbers in the bars correspond to Pro-Code identifications.
Mice were sacrificed 14 days after cell injection, and 18 different tumors were removed, and cultured for 3 days to enrich for the cancer cells. The cells were stained for NGFR and each of the 10 Pro-Code epitopes. We were able to identify 118 – 120 Pro-Code populations in each tumor (Figure 2B). While the proportion of each population varied for different Pro-Codes, this reflected a bias in the original population, as indicated by the comparison of each Pro-Code’s frequency in the pre-inoculation cells versus the tumors. Importantly, there was no significant difference in the proportion of the vast majority of Pro-Code populations in WT or Rag1−/− mice. This demonstrates the Pro-Codes are not differentially rejected, and can be used in vivo.
Although each mouse was injected with the same pool of cells, the specific Pro-Code composition of each tumor was different (Figure 2C). While most individual Pro-Codes were present in <1% of tumor cells, there was variability in the percent of each Pro-Code between tumors and mice. For each tumor, we plotted the proportion of the 10 most abundant Pro-Codes (Figure 2D). The same initial mix of 120 Pro-Code subpopulations developed into heterogenic tumors, in which 10 populations accounted for up to 50% of the total cell number. Some Pro-Code populations were abundant in every tumor (e.g. Pro-Codes 108 and 21), but their proportion within each tumor varied greatly, whereas other Pro-Code populations were only abundant in a single tumor, such as Pro-Code 6 (Figure 2B). These results support a model in which clonal growth was largely stochastic and not impacted by the Pro-Codes, and demonstrate Pro-Codes can be used for cell tracking studies.
Pro-Codes allow for high-dimensional phenotyping of CRISPR screens with single cell resolution
One of the advantages of the Pro-Codes is that it could permit addition of protein-level phenotyping in genetic screens. To test this possibility, we generated 96 CRISPR gRNAs targeting 54 different genes (1–3gRNA/gene), and paired each gRNA with a different Pro-Code. As it has recently been reported that packaging vector pools together can lead to varying degrees of barcode swapping (Hill et al., 2018; Sack et al., 2016), we made each vector individually, and subsequently pooled them in equimolar ratio, as this eliminates the possibility of swapping (Adamson et al., 2016). THP1 human monocytes were engineered to express Cas9 (THP1-Cas9), and transduced with all 96 Pro-Code/CRISPR vectors together as a pool. The cells were cultured for 10 days, then stained for NGFR, the Pro-Codes, and CD4, CD40, CD44, CD45, CD116, CD164, CD220, HLA-A, HLA-DR and IFNGR1, which were all targeted by CRISPR gRNAs in the vector library (Figure 3A). We then analyzed 500,000 cells by CyTOF. All 96 Pro-Code populations were resolved and clustered. This enabled us to examine expression of the surface proteins on each of the 96 Pro-Code/CRISPR populations with single cell resolution.
Figure 3. High content phenotypic analysis of monocytic cells engineered with a Pro-Code/CRISPR library.
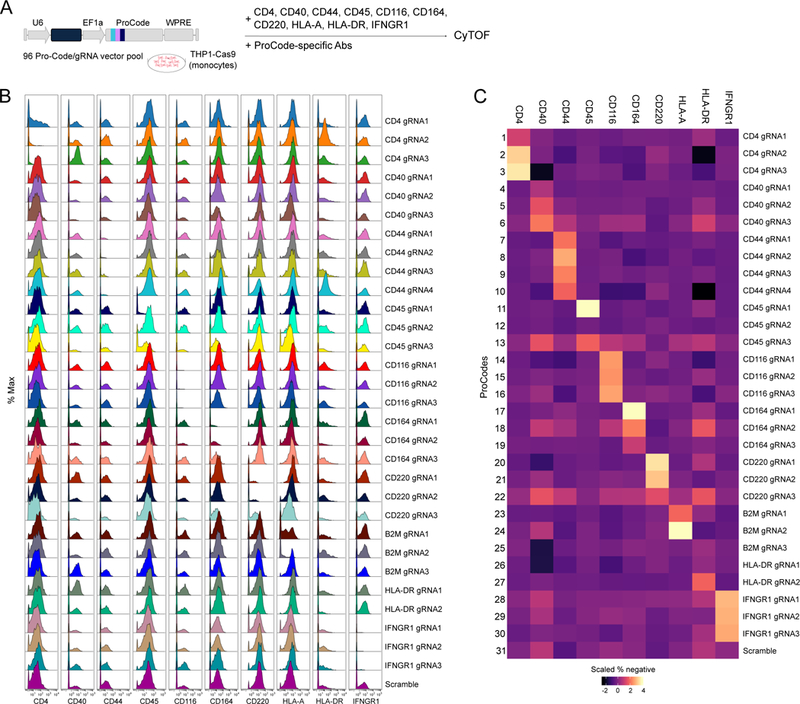
(A) Schematic of the Pro-Code/CRISPR phenotypic analysis. (B) Expression of the indicated proteins on each Pro-Code/CRISPR cell population. Shown are representative histograms for each Pro-Code population. The Y axis represents cell count normalized by protein detection channel. (C) Heatmap representation of the relative percent of protein negative cells for each Pro-Code population. All data is representative of 2 independent experiments.
In each Pro-Code population in which one of the membrane-bound proteins was targeted, there was an increase in the percent of cells negative for the cognate protein (Figure 3B, C). For example, in cells expressing Pro-Code 3, which was linked to a gRNA targeting the CD4 gene, 85% of the cells were CD4 negative, whereas cells expressing Pro-Codes linked to gRNAs targeting unrelated genes were almost all CD4 positive (Figure 3B, C and S4A). High efficiency protein loss was also observed for CD44, CD45, CD116, CD164, CD220, and IFNGR1. HLA-A (MHC class I) was expressed by >90% of cells in each Pro-Code/CRISPR cluster, except those expressing Pro-Codes 23 and 24, which were linked to gRNAs targeting B2m; in these populations 45% and 80% of THP1 were HLA-A negative, respectively. As B2m is required for HLA stability, the loss of HLA in these clusters represents a downstream phenotype of B2m KO. There was little evidence of KO for some gRNAs, consistent with the known variability in CRISPR efficiency between gRNAs. These results demonstrate Pro-Codes can mark cells encoding a specific CRISPR gRNA. They also demonstrate how Pro-Codes enable protein-level phenotyping in pooled CRISPR screens.
The library used above was made with vectors packaged individually and pooled subsequently. This prevents the possibility of barcode swapping. Recently, it was reported in pre-print studies that swapping can be reduced by co-packaging libraries with a low homology transfer vector (Adamson et al., 2018; Feldman et al., 2018). To determine if this would be compatible with the Pro-Codes, we produced the 96 Pro-Code/CRISPR library as a pool and during vector packaging we spiked in a plasmid encoding an LV expressing GFP and no CRISPR or Pro-Code. THP1-Cas9 cells were transduced with the 96 Pro-Code/CRISPR library at low MOI. The cells were stained for NGFR, the Pro-Code epitopes, and all 10 membrane-bound molecules, as above. We also stained for GFP to distinguish cells transduced with the GFP encoding LV in the pool, and analyzed cells by CyTOF. Similar to the library made with individually packaged vectors, we resolved all 96 Pro-Code populations, and consistently observed loss of a specific protein on a high percent of cells expressing a Pro-Code linked to a gRNA targeting the cognate gene (Figure S4B-C). The frequency of cells negative for the targeted protein was ~90% similar between the libraries generated with vectors produced individually or as a pool with the low homology spike in vector. These results indicate Pro-Code/CRISPR libraries can be produced as a pool and function at high efficiency, and further support the ability of Pro-Codes to facilitate high-dimensional phenotypic screens.
Pro-Codes enable interrogation of signaling pathways in reverse genetic screens
Intracellular signaling plays an essential role in numerous cellular processes. The activation and deactivation of specific proteins in signaling pathways is a post-translational event, and is thus optimally studied at the protein level. This makes it challenging to directly assess signaling alterations with current screening approaches. We decided to test if the Pro-Codes would facilitate a genetic screen of STAT signaling. STAT proteins function downstream of cytokine receptors. When different cytokines engage their cognate receptors, specific STAT proteins are phosphorylated, and transmit the cytokine signal. IFNγ engagement of the IFNγ receptor (comprised of IFNGR1 and IFNGR2 subunits) triggers phosphorylation of STAT1 (pSTAT1), whereas IL-6 induces pSTAT1 and pSTAT3, and GM-CSF induces pSTAT5 (Figure 4A, B).
Figure 4. Analysis of phospho-STAT signaling in Pro-Code/CRISPR engineered cells.
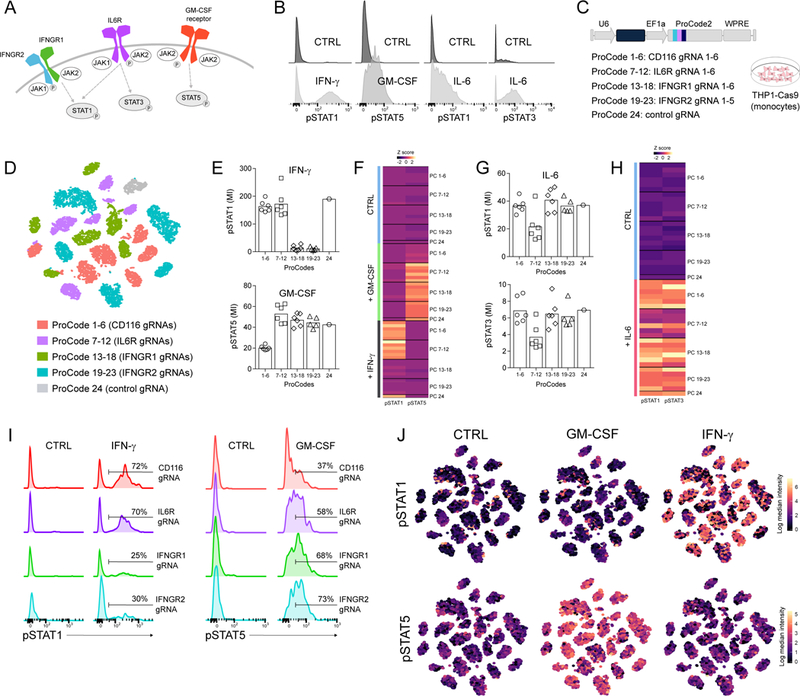
(A) Schematic overview of phospho-signaling downstream of the IFNγ, GM-CSF (CD116) and IL-6 (CD126) receptors. (B) THP1-Cas9 were stimulated with IFNγ, GM-CSF, IL-6 or PBS (ctrl), stained for pSTAT1, pSTAT3 and pSTAT5, and analyzed by CyTOF. Representative histograms shown (n=3 independent experiments). (C) Schematic of the ProCode/CRISPR library used in (D-J). (D) THP1-Cas9 were transduced with the 24 Pro-Code/CRISPR library, stimulated with the indicated cytokine, and analyzed for the Pro-Codes and pSTAT1 and pSTAT3 by CyTOF. Shown is the viSNE visualization of 24 Pro-Code/CRISPR populations colored by the target gene. (E) Expression of pSTAT1 and pSTAT5 in each Pro-Code population after GM-CSF or IFNγ. Bar plots present mean intensity (MI). Each point is a different Pro-Code/gRNA. (F) Relative expression of pSTAT1 and pSTAT5 across all CRISPR/Pro-Code populations after GM-CSF or IFNγ. (G) Expression of pSTAT1 and pSTAT3 in each Pro-Code population after IL-6. Bar plots present MI. (H) Relative expression of pSTAT1 and pSTAT3 across all CRISPR/Pro-Code populations after IL-6. (I) Levels of pSTAT1 and pSTAT5 after IFNγ and GM-CSF, respectively, in different ProCode/CRISPR populations; representative histograms shown. Y axis represents relative cell count. (J) viSNE visualization of pSTAT1 and pSTAT5 levels after GM-CSF or IFNγ. The Pro-Code/CRISPR identity of each cluster can be found in (D). Data is representative of 3 independent experiments.
We constructed a library of 24 LVs, each encoding a different Pro-Code and gRNA (Figure 4C). The gRNAs targeted the IFNGR1, IFNGR2, IL6R (IL-6 receptor), and CD116 (GM-CSF receptor) genes. We generated 5–6 gRNAs/gene, as well as one control gRNA targeting an irrelevant gene, and cloned each one with a different Pro-Code. THP1-Cas9 cells were transduced with the pool of Pro-Code/CRISPR vectors. After 1 week the cells were stimulated with IFNγ, GM-CSF, IL-6, or PBS. After 15 minutes cells were fixed, stained with metal-conjugated antibodies specific for the Pro-Code epitopes as well as pSTAT1, pSTAT3, and pSTAT5, and analyzed by CyTOF. All 24 Pro-Code populations were resolved and uniquely clustered (Figure 4D, S5A).
We examined the expression of pSTAT1, pSTAT3, and pSTAT5 in each Pro-Code population. In all cases, we observed decreased phospho-signaling in cells expressing a Pro-Code linked to a gRNA targeting the cognate receptor (Figure 4E-H, S5B). Looking at the mean change in signaling, there was a 15-fold decrease in pSTAT1 levels in cells expressing Pro-Codes linked to gRNAs targeting IFNGR1 and IFNGR2 (Figure 4E, F). Whereas in cells expressing the same Pro-Code/CRISPRs, pSTAT5, and pSTAT1 and pSTAT3 levels were normal in response to GM-CSF and IL-6. This indicated the IFNGR1 and IFNGR2 gRNAs only impaired pSTAT1 signaling in response to IFNγ. Similarly, in cells encoding the Pro-Codes linked to gRNAs targeting GM-CSF there was a 3-fold reduction in pSTAT5 levels in response to GM-CSF, and in cells carrying gRNAs targeting IL6R there was a 2-fold reduction in both pSTAT 1 and pSTAT3 levels in response to IL-6 (Figure 4G, H).
The ability to analyze the cells at single cell resolution enabled us to look at the heterogeneity in each Pro-Code/CRISPR population of cells. When we treated cells with IFNγ, 70% of the cells in the Pro-Code clusters linked to gRNAs targeting CD116 and IL6R had increased pSTAT1, whereas in the Pro-Code clusters linked to gRNAs targeting IFNGR1 and IFNGR2, only ~25% of the cells had increased pSTAT1 (Figure 4I, J). When the cells were treated with GM-CSF, 60–70% of the cells in the clusters encoding gRNAs targeting IL6R, IFNGR1, and IFNGR2 upregulated pSTAT5, but only 30–40% of the cells in the Pro-Code clusters encoding CD116 gRNAs upregulated pSTAT5 (Figure 4I, J).
Looking at the viSNE clusters, in which each dot is representative of a single cell, there were cells positive and negative for pSTAT (Figure 4J, S5). Thus, while the bulk analysis indicated a major reduction in pSTAT signaling downstream of the receptor targeted by a specific CRISPR, single cell analysis indicated there was significant heterogeneity between cells even within the same Pro-Code cluster. This heterogeneity reflects biological differences between cells in their response to cytokine stimulation, but also reveals cell-to-cell heterogeneity in CRISPR KO, as observed in our studies above measuring the protein levels of the gene targeted by specific CRISPRs. This is not unexpected, as the editing efficiency of CRISPR is variable, but highlights the important utility of single cell analysis in CRISPR screens. Together, these results demonstrate Pro-Codes enable direct single cell phenotypic analysis of signaling pathways in CRISPR screens, which is not feasible with DNA or RNA level analysis.
Pro-Code/CRISPR screen reveals mechanisms of cancer resistance to antigen-specific cytotoxic T cells
Cancer cells acquire mutations, which generate neo-antigens that are loaded on to MHC class I (MHC-I), and make the cancer cells targets for CD8+ T cell killing (Schumacher and Hacohen, 2016). However, cancer cells can alter gene expression to resist being killed. Though some of the genes important for cancer immune editing have been identified, the potential contributions of many genes still need to be interrogated. Recently, several studies have performed pooled CRISPR screens, using DNA barcodes for deconvolution, to identify novel sensitivity and resistance genes (Manguso et al., 2017; Pan et al., 2018; Patel et al., 2017). We set out to determine if we could use the Pro-Codes to aid in the identification of genes conferring cancer cell sensitivity or resistance to T cell immunity.
We generated a library of 56 CRISPR gRNAs targeting 14 different genes (3 to 4 gRNAs/gene), and paired each CRISPR with a unique Pro-Code, to form a pool of 56 Pro-Code/CRISPR vectors (including 4 scrambled gRNAs) (Figure 5A). We selected the 14 genes to contain known regulators of immunity, such as B2m, and several genes with no known role, such as Cldn4. As a model of breast cancer, we utilized the 4T1 mammary carcinoma line. In previous screens, antigen-specific T cells targeting model tumor associated antigen (TAA), such as OVA, gp100, and NY-ESO-1, were used (Manguso et al., 2017; Pan et al., 2018; Patel et al., 2017). A caveat of these antigens is they are not readily detected in cells. To overcome this limitation, we took advantage of just eGFP death inducing (Jedi) T cells, which express a T cell receptor that recognizes the immunodominant epitope of GFP loaded in the H-2Kd allele of MHC-I (Agudo et al., 2015). Jedi T cells enable GFP to be used as a model antigen that can be easily detected (Agudo et al., 2018). We engineered 4T1 to express either GFP (4T1-GFP) or near-infrared fluorescent protein 670 (4T1-RFP) alone, or with Cas9 (4T1-Cas9-GFP and 4T1-Cas9-RFP). When the cells were co-cultured with activated CD8+ Jedi T cells there was selective killing of the GFP+ cells, which could be quantified by flow cytometry (Figure 5B, C). Thus, this system enables precise analysis of antigen-specific T cell killing. The RFP+ cells serve as an internal control of non-TAA expressing cells, and enables distinction between the effects of a specific KO on cell fitness versus T cell sensitivity.
Figure 5. Pro-Code/CRISPR screen for genes conferring sensitivity or resistance to antigen-dependent T cell killing.
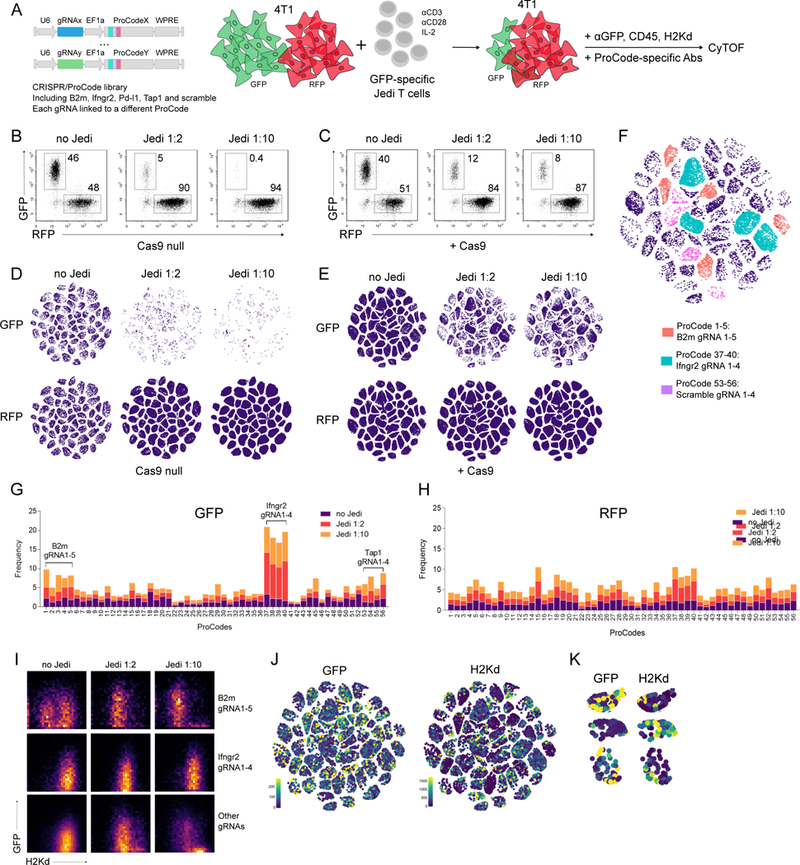
(A) Schematic of the immune editing co-culture system and the ProCode/CRISPR library. 4T1 cells (+/−Cas9, +/−GFP/RFP) were transduced with a library of 56 ProCode/CRISPR vectors, co-cultured with activated Jedi T cells, and analyzed by CyTOF. (B) Frequency of GFP+ and RFP+ 4T1 cells was measured by flow cytometry. Representative dotplots are shown. Jedi 1:2 and Jedi 1:10 is 2-fold and 10-fold multiple of T cells to cancer cells, respectively. (C) Frequency of GFP+ and RFP+ 4T1-Cas9 cells was measured by flow cytometry. Representative dotplots are shown. (D) viSNE visualization of the 4T1-GFP and 4T1-RFP Pro-Code populations co-cultured alone or with activated Jedi T cells. Each cluster corresponds to a different Pro-Code. (E) viSNE visualization of the 4T1-GFP-Cas9 and 4T1-RFP-Cas9 Pro-Code populations co-cultured alone or with activated Jedi T cells. Each cluster corresponds to a different Pro-Code. (F) viSNE visualization of 56 Pro-Code/CRISPR populations (GFP-4T1-Cas9, Jedi 1:10) colored by the target: orange=B2m, cyan=Ifngr2, purple=scramble, navy=others. (G, H) Frequency of each Pro-Code/CRISPR populations among the GFP-4T1-Cas9 (G) and RFP-4T1-Cas9 (H) cells in the absence (no Jedi) or presence (Jedi 1:2, Jedi 1:10) of GFP- specific Jedi T cells. (I) GFP and H2Kd expression on 4T1-Cas9-GFP cells expressing gRNAs targeting B2m, Ifngr2 and all other genes. (J) GFP and H2Kd expression levels Pro-Code/CRISPR populations in GFP-4T1-Cas9 cells resisting T cell killing (Jedi 1:10); (K) GFP and H2Kd expression on selected Pro-Code populations (from J). Data is representative of 3 independent experiments.
We transduced each group of 4T1 (4T1-GFP, 4T1-RFP, 4T1-Cas9-GFP, and 4T1-Cas9-RFP) with the Pro-Code/CRISPR library. After 10 days, 4T1-Cas9-GFP and 4T1-Cas9-RFP (or 4T1-GFP and 4T1-RFP) cells were mixed in a 1:1 ratio, and co-cultured with activated CD8+ Jedi T cells (Figure 5A). Bulk comparison indicated GFP+ cells were almost completely eliminated in Cas9 null cultures with activated Jedi T cells (Figure 5B). In contrast, a large fraction of 4T1-Cas9-GFP cells survived (8–12% of the culture), despite their expression of the antigenic target of the T cells (Figure 5C). These results suggested gene editing was resulting in resistant cancer cells.
To determine which genes may be involved in 4T1 resistance to T cell killing, we stained the cells with metal-conjugated antibodies for the Pro-Code epitopes, as well as GFP, CD45 and MHC-I (H-2Kd), and analyzed by CyTOF. Each of the 56 Pro-Code populations were detected (Figure 5D, E). There were no changes in the relative frequency of specific Pro-Code populations in 4T1-RFP cells, with or without Cas9, in the presence or absence of Jedi T cells (Figure 5D, E, lower panels). Examination of the Pro-Code markers in the surviving 4T1-Cas9-GFP population revealed enrichment of cells expressing Pro-Codes linked to gRNAs targeting Ifngr2 and B2m (Figure 5E-H). Approximately 39% of the surviving cancer cells carried an Ifngr2 CRISPR (Figure 5G). We saw a similar result when we performed the experiments with individual CRISPRs targeting only B2m or Ifngr2 (Figure S6A). These findings are consistent with emerging clinical data correlating resistance to checkpoint inhibitors with mutations in the B2m and IFNγ pathways (Gao et al., 2016; Zaretsky et al., 2016), and with recent genome-wide CRISPR screening data (Patel et al., 2017).
Because the Pro-Codes allowed us to perform the analysis at the protein level and with single cell resolution, we could also examine the expression of both the TAA (GFP) and MHC-I on each cell. As expected, we could detect lower MHC-I on cells encoding the B2m gRNAs (Figure S6B). In cells encoding Ifngr2 CRISPRs, there were normal levels of MHC-I expression in steady-state, but the expression of MHC-I on these cells did not increase in the Jedi co-cultures, as it did in cells carrying unrelated CRISPRs. This suggests one of the mechanisms by which the Ifngr2 CRISPR cells resisted T cell killing may be due to diminished upregulation of MHC-I.
In addition to the B2m and Ifngr2 CRISPR populations, there were residual cells remaining in each Pro-Code/CRISPR population after Jedi co-culture (Figure 5D, E). Interestingly, a common feature of these cells was decreased gFp and/or MHC-I (Figure 5I-K and S6C). Since it is possible some of the H-2Kdlow cells could have resulted from a B2m gRNA swapping in to another Pro-Code vector, we performed the same experiment with individual Pro-Code/CRISPR vectors encoding a scrambled gRNA. As we observed with the pool of vectors, in cultures containing activated Jedi T cells there emerged populations of 4T1-GFP that had downregulated H-2Kd or GFP and escaped T cell killing (Figure S6D), supporting the notion that this mechanism can arise spontaneously.
The IFNγ inducible genes Psmb8 and Rtp4 influence susceptibility to antigen-dependent T cell killing
Though the cells carrying the Ifngr2 CRISPR did not upregulate MHC-I in response to IFNγ, the cells still expressed high levels of MHC-I (Figure S6B). Indeed, the levels of MHC-I were comparable to the activated Jedi T cells. Since there are many facets of the IFNγ pathway, we decided to look at what other components of the pathway may influence cancer resistance to T cell killing. We selected genes associated with the IFNγ pathway, as well as several genes with no reported associations (Socs1–7, Ptpn1, Ptpn2, Rtp4, Rab5b, Stip1, Supt16, and Psmb8). We designed 2–4 gRNAs per gene, and cloned each one into a Pro-Code construct. A pool of 56 Pro-Code/CRISPR LVs were generated, and used to transduce 4T1-GFP-Cas9 and 4T1-Cas9-mCherry cells. The transduced populations were mixed in a 1:1 ratio, and co-cultured with or without activated Jedi T cells. On day 3, the cells were collected and stained with metal-conjugated antibodies for the Pro-Code epitopes, as well as GFP, mCherry, CD45, MHC-I (H-2Kd) and PD-L1, and analyzed by CyTOF.
Bulk comparison of GFP+ and mCherry+ cells found that a fraction of GFP+ cells survived, indicating resistant cancer cells had emerged (Figure 6A). As expected, cells exposed to activated Jedi T cells upregulated both MHC-I and PD-L1 (Figure 6B, C). Interestingly, when we looked at PD-L1 expression on specific Pro-Code populations, all 3 populations expressing a Pro-Code linked to a gRNA targeting Socs1 had increased upregulation of PD-L1 (Figure 6D). This was specific to PD-L1 because the same population of cells had similar levels of MHC-I to other Pro-Code/CRISPR populations (Figure 6E). These results implicate Socs1 as a negative regulator of PD-L1.
Figure 6. Pro-Code/CRISPR analysis of select IFN𝛄-inducible genes in cancer cell killing by antigen-specific T cells.
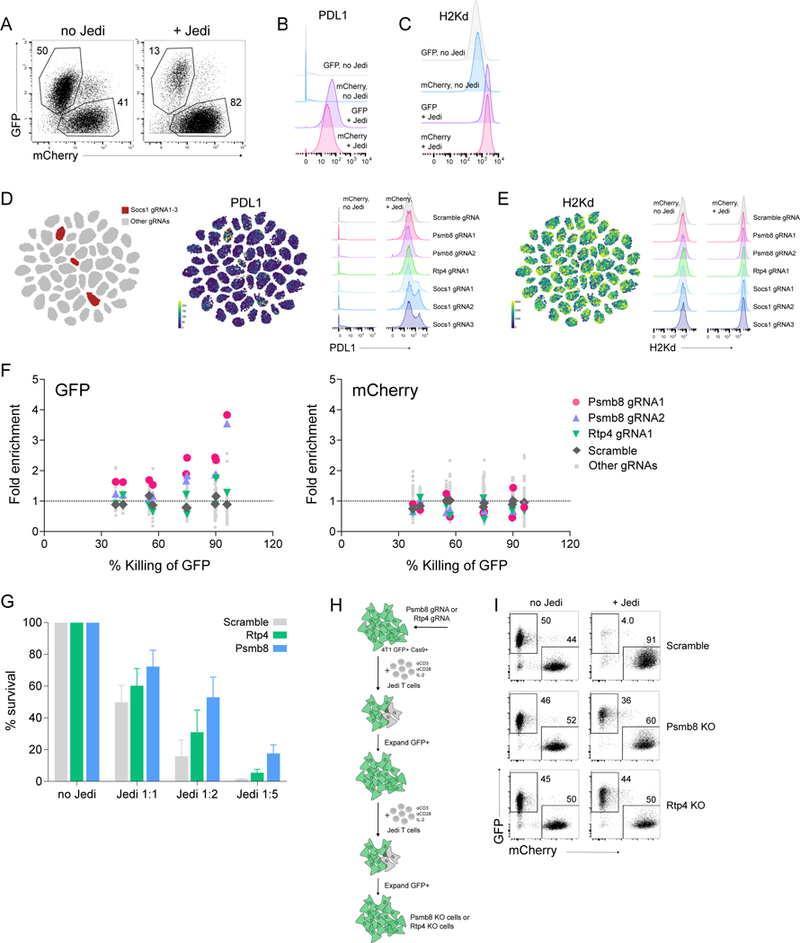
(A-F) 4T1-Cas9-GFP and 4T1-Cas9-mCherry cells were transduced with 56 ProCode/CRISPR vectors, mixed in a 1:1 ratio, and co-cultured with activated Jedi T cells. On day 3, cells were collected, stained for the Pro-Code, as well as GFP, mCherry, CD45, H2Kd and PD-L1, and analyzed by CyTOF. (A) Frequency of cells were measured by CyTOF; no Jedi - no T cells added, + Jedi - 4-fold excess of T cells over cancer cells. Representative dotplots shown. (B) PDL1 (C) H2Kd expression in the bulk GFP+ and mCherry+ cell populations. (D, E) viSNE visualization and histograms showing PDL1 (D) and H2Kd (E) expression on individual Pro-Code/CRISPR populations among mCherry+ cells. (F) Fold enrichment of Psmb8, Rtp4 and scramble Pro-Code/CRISPR populations (+ Jedi vs. no Jedi conditions) shown as a function of % killing by Jedi T cells. Each point is from an independent experiment with two different ratios of Jedi to cancer cells. 4 independent experiments were performed. (G) GFP-4T1-Cas9 cells were transduced with gRNAs targeting Psmb8, Rtp4 or scramble gRNA. The frequency of GFP+ cells in the absence (no Jedi) or presence (Jedi 1:1, Jedi 1:2, Jedi 1:5) of Jedi T cells was determined by flow cytometry. Bar graphs present the mean±SD (n = 3). 4T1-Cas9-mCherry cells were used as control. Note that the percent of surviving cells is dependent on CRISPR knockout efficiency, and is thus not quantitative, as indicated by (I). (H) Schematic overview of the Psmb8 and Rtp4 validation approach. (I) 4T1-Cas9-GFP cells transduced with a vector encoding a Psmb8, Rtp4, or scramble gRNA were selected as shown in (H) and mixed with activated Jedi T cells, and cultured for 3 days. Frequency of GFP+ and mCherry+ cells in the absence (no Jedi) or presence (+ Jedi) of Jedi T cells is shown. Dotplots are representative of 2 independent experiments.
Next, we analyzed changes in the frequency of specific Pro-Code populations within the GFP and mCherry cell fractions (Figure 6F). To allow for comparison across 4 independent experiments, we expressed these changes as a function of killing of the GFP+ cells. Examination of the Pro-Code markers revealed that cells expressing Pro-Codes linked to gRNAs targeting Psmb8 and Rtp4 were enriched in the surviving 4T1-Cas9-GFP populations. The frequency of 4T1-Cas9-mCherry cells expressing Psmb8 and Rtp4 gRNAs did not significantly change, indicating enrichment was dependent on antigen-specific T cell killing.
To validate our findings, 4T1-Cas9-GFP cells were transduced with either gRNAs targeting Psmb8 or Rtp4, or a scramble gRNA, mixed in 1:1 ratio with 4T1-Cas9-mCherry cells, and co-cultured with activated CD8+Jedi T cells. In support of the screen results, we observed increased resistance of cells encoding the Psmb8 and Rtp4 CRISPR compared to the scramble control (Figure 6G and S7A). Whereas <0.1% of control 4T1-GFP cells remained in the Jedi co-cultures, ~4% of the Rtp4 CRISPR and 10% of the Psmb8 CRISPR 4T1-GFP cells remained.
Though not all transduced cells were resistant, this is expected because not all cells will be a complete KO for Rtp4 or Psmb8, due to variability in CRISPR efficiency. Thus, the percent of cells remaining reflects resistance to T cell killing, but does not provide an indication of the robustness of resistance. To address this, we co-cultured 4T1-Cas9-GFP cells expressing the Rtp4 or Psmb8 gRNA with activated Jedi T cells, and expanded the GFP+ resistant cells (Figure 6H). The cells were mixed with 4T1-Cas9-mCherry cells, and recultured with activated Jedi T cells. Strikingly, the Psmb8 and Rtp4 KO cells were almost completely resistant to T cell killing (Figure 6I). Western blot confirmed Psmb8 protein was absent in the expanded Psmb8 CRISPR 4T1 cells (Figure S7B). Because there was not a satisfactory antibody for Rtp4 protein detection, we used Sanger sequencing and qPCR and confirmed the Rtp4 gene had been mutated and was no longer expressed (Figure S7C,D). Together, these results indicate Psmb8 and Rtp4 have a non-redundant role in mediating sensitivity of tumor cells to antigen-dependent T cell killing.
Discussion
This work describes a new technology for cell and vector barcoding, which uses combinations of linear epitopes to create a higher multiple of protein barcodes. We successfully generated and resolved 364 unique Pro-Codes using 14 epitope and antibody pairs for construction and detection. While this is far fewer barcodes than achieved with DNa, it is an order of magnitude greater than what currently exists with protein reporters. Moreover, 1000s of new Pro-Codes can be created by applying the principle we used here and introducing additional epitopes and epitope positions. Though generating genome-wide Pro-Code/CRISPR libraries cannot be done at the relative ease of DNA barcoded libraries, the Pro-Code’s will primarily be for more focused screens; concentrating on specific pathways or gene classes, and targeting 100–500 genes. As more epitopes are validated, it will also be possible to create Pro-Code/CRISPR libraries with non-overlapping epitopes, and use them together in complex screens to identify cooperating or redundant genes in a relatively unbiased manner.
An important advance provided by the Pro-Codes is the ability to perform high-dimensional phenotyping of multiple proteins in pooled screens. This was not feasible with DNA barcodes, as the screen readout was limited to measuring changes in barcode frequency, and inferring phenotype based on the selective pressure applied. By being able to mark 100s of different CRISPR-expressing populations and measure many protein markers, Pro-Codes expand the types of pooled genetic screens that can be performed, and will help facilitate the annotation of gene functions.
A key feature of the Pro-Codes is that they enable screens to be performed with single cell resolution. For CRISPR screens, single cell analysis is particularly relevant because the efficiency of CRISPR KO is highly variable; some cells may be complete KO, while other cells have only a partial KO or remain wildtype. This was evident from our phenotypic analysis in which only a fraction of cells expressing a particular Pro-Code/CRISPR were negative for the cognate protein. As DNA barcode de-convolution is generally performed on bulk cells, this means cells with complete, partial or no KO are lumped together in the analysis. Even if there is an effect of complete KO, the magnitude is diluted by the wildtype cells. With Pro-Codes, every cell expressing a CRISPR is analyzed individually. Even when the targeted gene itself is not analyzed, the phenotypic differences can be seen between individual cells receiving the same CRISPR, as we observed in the Pro-Code/CRISPR analysis of phospho-STAT signaling, as well as PD-L1. Moreover, as opposed to DNA barcodes in which the percent of each vector is presumed from sequence frequency, with the Pro-Codes, the frequency of each CRISPR-carrying cell within a population is directly determined. This enables precise consideration of the number of cells sampled in each population and informs analysis.
Several groups have incorporated scRNA-seq into pooled screens to obtain more comprehensive phenotyping than previously possible, and to achieve single cell resolution (Adamson et al., 2016; Datlinger et al., 2017; Dixit et al., 2016; Jaitin et al., 2016). This provides a powerful advance to pooled screening. However, the cell throughput of scRNA-seq is still relatively limited compared to what can be achieved with CyTOF (thousands versus millions), and the efficiency of transcript capture makes it challenging to quantitatively compare gene expression on a per cell basis without imputing gene levels. As gene editing does not necessarily affect the level of a target transcript, it is also difficult to directly determine if there is functional KO of a particular gene by scRNA-seq. The Pro-Codes makes it possible to analyze millions of single cells with precise quantification of protein levels. Though the number of genes that can be analyzed by CyTOF is fewer than scRNA-seq, it should be feasible to expand the phenotyping space using oligo-labeled antibodies to detect the Pro-Codes and other proteins, and to deconvolute with single cell sequencing, as has been described (Peterson et al., 2017; Stoeckius et al., 2017).
As noted, barcode swapping can occur in retroviral vector libraries packaged as pools, and the degree of swapping can range from 6% to 50%, depending on the distance between the barcode and effector molecule (i.e. gRNA, shRNA, or cDNA) (Hill et al., 2018; Sack et al., 2016). Swapping occurs when two different vector genomes are packaged in the same virion, and there is template switching during reverse transcription (King et al., 2008). Fortunately, swapping can be prevented by packaging each vector individually, and pooling them subsequently, as we did in our library targeting surface proteins. We also found that introducing a plasmid encoding an LV with low-homology to the library during vector production reduces barcode swapping, as the efficiency of KO was similar between Pro-Code/CRISPR libraries made with individually packaged vectors or vectors produced as a pool with the low-homology vector. This works by co-packaging a genome in the vector particle that does not serve as a suitable partner for swapping with the library genome, and was also reported to be effective in pre-print studies (Adamson et al., 2018; Feldman et al., 2018). The advantage is that it permits the CRISPR libraries to be made as a pool, which makes it more feasible to produce larger libraries.
In this study, we utilized CyTOF for Pro-Code detection because it enabled concurrent detection of many proteins. It should be possible to detect the Pro-Codes by flow cytometry, and this could be used to sort particular Pro-Code populations for expansion and further study. There is also the potential to utilize the Pro-Codes with histological techniques, and add spatial mapping to CRISPR screens. There are now at least two platforms that enable high-dimensional tissue imaging with antibodies, allowing over 40 parameters to be simultaneously detected in a single section (Angelo et al., 2014; Giesen et al., 2014). This would enable each of the Pro-Code epitopes to be detected, and thus hundreds to thousands of barcoded cells to be resolved in a tissue section, along with >30 different protein markers of cell identity and function.
We used the Pro-Codes to carry out CRISPR screens aimed at identifying genes that influence sensitivity to antigen-specific T cell killing. The screens were primarily intended as proof-of-principle studies, and were thus relatively small and included genes with established importance, such as B2m and Ifngr2. The IFNγ pathway has been implicated as a key component in the clinical response to checkpoint inhibitors (Minn and Wherry, 2016). Mutations in IFNGR1 and JAK, a component of the IFNγ signaling pathway, have been found in patients presenting resistance to checkpoint inhibitors (Gao et al., 2016; Zaretsky et al., 2016). However, the mechanisms that make IFNγ signaling essential to immune editing are not well established. Our studies found KO of two IFNγ inducible genes, Psmb8 and Rtp4, resulted in resistance to antigen-specific T cell killing. Psmb8 (also known as Lmp7) is a component of the immunoproteasome, which functions in generating peptides for MHC-I (Basler et al., 2013), and its expression has been found to positively correlate with tumor-infiltrating lymphocyte abundance in breast cancer (Lee et al., 2018). Rtp4 (Receptor transporter protein 4) is a chaperone protein involved in folding G protein coupled receptors (Decaillot et al., 2008). The only defined targets of Rtp4 are opioid receptors (Decaillot et al., 2008), and, despite being an IFN stimulated gene, almost nothing is known about the role of Rtp4 in immunity. Future studies will be needed to understand how Rtp4 influences sensitivity to T cell killing, and to determine its relevance to immune editing of patient tumors.
The importance of analyzing phenotypic markers in the screen was highlighted by the discovery that many resistant cells had lower levels of MHC-I or the target antigen, GFP. While it is not surprising that loss of antigen or MHC-I would enable cancer cells to resist killing by antigen-specific T cells, our studies found that downregulation, and not just loss, of either factor also provided a survival advantage. This may be underappreciated as a mechanism of cancer resistance to cytotoxic T cell clearance, as subtle reductions in the expression of neo-antigens on individual cancer cells has not been widely examined in tumors owing to the challenge of making these measurements. Understanding the quantitative relationship between presentation components, neoantigen levels, and the immunotherapy response at high resolution in patient’s tumors is needed, especially as neo-antigen prediction and neo-antigen vaccines (Ott et al., 2017) become more widely used in cancer immunotherapy.
Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian D. Brown (brian.brown@mssm.edu).
Experimental Model and Subjects Details
Mice
BALB/c and BALB/c Rag1−/− mice were purchased from Jackson Laboratory. Jedi mice (Agudo et al, 2015) were from our own established colonies. All mice were hosted in a specific pathogen-free facility. At the time of experimentation, mice were 8–12 weeks of age. The Institutional Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai reviewed and approved all protocols for animal usage described in the present study.
Cell culture
293T cells were grown in IMDM with 10% heat-inactivated FBS (Gibco), 100 U/ml penicillin/streptomycin (Gibco) and 2 mM L-Glutamin. Cells were passaged up to 20 times (washed with PBS, detached from the plate with 0.05% Trypsin-EDTA (Gibco) and replated). After 20 passages, cells were discarded. THP-1 were grown in DMEM with 10% heat-inactivated FBS (Gibco), 100 U/ml penicillin/streptomycin (Gibco), 2 mM L-Glutamin and 55 uM 2-mercaptoethanol. Jurkat cells were grown in RPMI with 10% heat-inactivated FBS (Gibco), 100 U/ml penicillin/streptomycin (Gibco) and 2 mM L-Glutamin. Cells were maintained at a maximum concentration of 1 million per ml. Both Jurkat and THP-1 cells were maintained at a maximum concentration of 1 million per ml. 4T1 cells are a BALB/c cell line of mammary carcinoma. They were cultured in RPMI with 10% heat-inactivated FBS (Gibco), 100 U/ml penicillin/streptomycin (Gibco) and 2 mM L-Glutamin. Cells were kept at a maximum confluency of 70% and passaged up to 20 times as described for 293T cells. All cell lines were purchased from ATCC.
Method Details
Vector construction
Linear epitope sequences were cloned into lentiviral vector downstream of the human EF1a promoter in the C terminal region of the dNGFR cDNA using ShpI and BsrGI restriction sites. The Pro-Code vector also contained a U6 gRNA expression cassette similar to the one present in pX330 plasmid (Cong et al., 2013). BbsI sites were present downstream of the U6 promoter and upstream of the Cas9 gRNA scaffold for efficient gRNA cloning. Linear epitope sequences were codon-optimized to facilitate expression in mammalian cell systems, organized in combinations of 3 and separated by a flexible linker comprised of six glutamines. Amino acid and nucleotide sequences of the epitope tags and the linker are provided in Table S1. To clone gRNA sequences, Pro-Code vectors were digested with BbsI, purified using pCr purification kit (Qiagen) and ligated with pairs of annealed oligo sequences (forward oligo design: 5’ CACCG(N)20; reverse oligo design: 5’ AAAC(N)20C, where (N)20 is the sequence of guide RNA or its reverse complement counterpart). sgRNA sequences were obtained from Brunello (human) or Brie (mouse) CRISPR libraries (Doench et al., 2016). TOP10 competent cells were used for all subsequent plasmid preparations with exception of lentiCRISPR v2 (Addgene plasmid no. 52961) (Sanjana et al., 2014), which was propagated using NEB stable competent cells (New England BioLabs). All plasmids were purified using ZR Plasmid Miniprep Classic kit (Zymo Research) or EndoFree Plasmid Maxi Kit (Qiagen).
Pro-Code/CRISPR libraries
The following genes were targeted in the Pro-Code CRISPR library used in Figure 3: B2M, CD116, CD164, CD220, CD4, CD40, CD44, CD45, HLADRA, IFNGR1, AKT1, AKT2, CBLB, CCR7, CD244, CD27, CD274, CD28, CD38, CD3E, CD62L, CTLA4, F8, FOS, FOSB, FOXO1, FOXO3, HAVCR2, ICOS, IFNGR2, IL2RA, IL2RB, IL2RG, IL7R, JUN, LAG3, MAP4K1, MAPK1, MAPK3, MAPK8, MAPK9, NFATC1, NFATC3, NFATC4, NFKB1, PDCD1, PRKCQ, STAT3, STAT5A, STAT5B, TIGIT, TNFRSF18, TNFRSF4 and ZAP70. The following genes were targeted in the Pro-Code CRISPR library used in Figure 5: B2m, Tap1, H2-D1, Pd-l1,Fak, Ccr4, Nlrc5, Cxcr7, Cd40, Ifngr2, Cldn4, Ephb2 and H2-Ke6. The following genes were targeted in the Pro-Code CRISPR library used in Figure 6: Socs1–7, Ptpn1, Ptpn2, Rtp4, Rab5b, Stip1, Supt16, and Psmb8.
Lentiviral vector production and titration
Lentiviral vectors were produced as previously described in detail (Baccarini et al., 2011). Briefly, 293T cells were seeded 24 hours before calcium phosphate transfection with third-generation VSV-pseudotyped packaging plasmids and the transfer plasmids. Supernatants were then collected, passed through a 0.22-μm filter, purified by ultracentrifugation, aliquoted and stored at −80˚C. Viral titer was estimated on 2 93T cells by limiting dilution. LentiCRISPR v2 transfer plasmid encoding Cas9 transgene and a puromycin resistant cassette was used to generate Cas9 lentivirus. To produce LV Pro-Code libraries, equimolar amounts of single plasmids were pooled and subsequently used for vector production. Alternatively, each LV was produced individually in a 96-well format, and all LVs were pooled in equimolar ratio before transduction. Where indicated, the Pro-Code libraries were co-transfected with pCCLsin.PPT.hPGK.GFP at 50% of total transfer plasmids.
Vector transduction
293T, THP-1, Jurkat and 4T1 cells were transduced as previously described (Mullokandov et al., 2012). To ensure that a majority of transduced cells received only one vector, fewer than 10% of cells were transduced in all experiments. For knockout experiments, THP1, Jurkat and 4T1 cells were engineered to stably express Cas9. Briefly, cells were seeded 24 hours prior to transduction in 6-well plates at 5–104 cells per well, and transduced with Cas9 lentivirus in the presence of 5 μg/ml polybrene (Millipore). 48 hours after transduction, cells were treated overnight with 10 ^g/ml puromycin (ThermoFisher) to remove all non-transduced cells. Puromycin treatment was repeated two additional times to ensure cell purity. Cas9 expression was confirmed by western blot using anti-Cas9 antibody (Millipore, clone 7A9). For T cell killing experiments, 4T1 cells (+/−Cas9) were first transduced with GFP, iRFP670 or mCherry lentiviral vectors, then with Pro-Code/CRISPR libraries.
Flow cytometry and cell sorting
Before FACS analysis, adherent cells were detached with 0.05% trypsin-EDTA, washed and resuspended in sterile PBS. Cells grown in suspension were washed and resuspended in sterile PBS. For analysis of NGFR, GFP or iRFP670 expression, cells were washed and resuspended in flow buffer (PBS, 2 mM EDTA, 0.5% BSA). For immune staining, flow buffer was supplemented either with anti-mouse CD16/CD32 antibody (eBioscience) or Human TruStain FcX Fc Receptor Blocking Solution (BioLegend). Following antibodies were used for flow analysis: anti-human CD271 PE and APC (BD Biosciences), anti-mouse H2Kd PE, Pacific Blue or biotin, anti-mouse B2m PE, anti-mouse CD45 PE-Cy7 (all from eBioscience), streptavidin PE-Cy7 (BioLegend). Data was acquired using BD Fortessa (BD) and analysis was performed using Cytobank or FlowJo Software (FlowJo, LLC). For T cell killing experiments, transduced 4T1 cells were sorted on a FACS Aria II (BD) to enrich for the NGFR+/GFP+, NGFR+/iRFP670+ or NGFR+/mCherry+ populations.
Tumor model
4T1 murine mammary gland carcinoma cells were injected (5×104 cells) in the mammary fat pad of 8–12 week old BALB/c WT or Rag1−/− mice. Tumor-inoculated mice were sacrificed 14 days later. Tumor cell suspensions were obtained by enzymatic treatment with RPMI supplemented with collagenase (1.5 mg/ml) and BSA (25 mg/ml) (45 min at 37°C). Digested tumors were homogenized by multiple passage through a 19G needle and filtered twice through a 40-μm cell strainer. Cells were put in culture with 6-thioguanine (60 μM) for 3 days to enrich for 4T1 cells, and remove stromal cells (hematopoietic, fibroblast, and endothelial) so that they would not be part of the cellular mixture analyzed. 3×106 cells per tumor were analyzed for Pro-Code distribution by CyTOF.
T cell killing assay
CD8+ T cells were isolated from spleens of Jedi mice. Splenic cells suspensions were obtained by mechanical disruption and filtering through 70-μm cell strainer. Red blood cells were lysed using RBC buffer (eBioscience), and CD8+ T cells were negatively selected using EasySep mouse CD8+ T cells isolation kit from StemCell Technologies, following manufacturer’s instructions. Cells were activated for 3 days with 5 μg/ml plate-bound anti-CD3 mAb (clone 2C11, BioXCell), 1 μg/ml anti-CD28 mAb (clone 37.51, BioXCell) and 20 ng/ml mouse recombinant IL-2 (Peprotech) in RPMI with 10% FBS, 100 U/ml penicillin/streptomycin, 2 mM L-glutamine, 1% non-essential amino acids, 1 mM sodium pyruvate 55 μM 2-mercaptoethanol and 20 mM HEPES. 4T1 cells (+/−Cas9, +/−GFP, +/−iRFP670 (Shcherbakova and Verkhusha, 2013), +/−mCherry) were transduced with the Pro-Code/CRISPR vector pool at a MOI of 1 and cell sorted based on NGFR expression. A 50:50 mix of GFP+ (target cells) and either iRFP670+ or mCherry (bystander cells) 4T1 cells were plated in 24-well plates (4 104 cells per well). Activated T cells were added to the wells 6 hour later, at different ratios. Cells were passaged every 2 days and seeded in a 6-well plate at day 2 and in a 10 cm dish at day 6. Killing was assessed by flow cytometry at day 2 and 4. At day 3 or 6, 3106 cells were stained with the antibodies specific for Pro-Code epitope tags, cD45, H2-Kd, PD-L1, mCherry and GFP and analyzed by CyTOF.
Mass cytometry
Antibodies were either purchased pre-conjugated from Fluidigm or purchased purified and conjugated in-house using MaxPar X8 Polymer Kits (Fluidigm) according to the manufacturer’s instructions. Following antibodies were used for CyTOF staining: HA tag-147Sm (clone 6E2, Cell Signaling), V5 tag-152Sm (Thermo Fisher Scientific), anti- DYKDDDDK (FLAG) tag-175Lu (clone 5A8E5, GenScript), VSVg tag-158Gd (rabbit pAb, Thermo Fisher Scientific), E tag-154Sm (clone 10B11, Abcam), E2 tag-160Gd (rabbit pAb, GenScript), NWSHPQFEK (NWS) tag-159Tb (clone 5A9F9, GenScript), S1 tag-153Eu (rabbit pAb, GenScript), AU1–162Dy (clone AU1, BioLegend), AU5–169Tm (clone AU5, BioLegend), H2Kd-biotin or H2Kd-149Sm (clone SF1–1.1.1, eBioscience), αGFP-155Gd (clone FM264G, BioLegend), αmCherry-142Nd (Abcam), anti-mouse CD274–149Sm (MiH5, eBioscience), anti-human CD126–151Eu (clone UV4, BioLegend), anti-human CD4 145 Nd (clone RPAT4, Fluidigm), anti-human CD40 164 Dy (clone 5C3, BioLegend), anti-human CD44 166 Er (clone BJ18, Fluidigm), anti-human CD45 115 In (clone HI30, BioLegend), anti-human CD116 165 Ho (clone 4H1, BioLegend), anti-human CD164 173 Yb (clone 67D2, BioLegend), anti-human CD220 149 Sm (clone B6.220, BioLegend), anti-human HLA-A,B,C 163 Dy (clone W6/32, BioLegend), anti-human HLA-DR 174 Yb (clone L243, Fluidigm), anti-human CD119-biotin (eBioscience), phospho STAT1–153Eu (Fluidigm), phospho STAT3 PE (eBioscience), phospho STAT5–150Nd (Fluidigm), anti-PE-165Ho, anti-biotin-143Nd (Fluidigm), anti-mouse CD90.2–113In (Fluidigm) and anti-mouse CD45–141pr (Fluidigm). Before CyTOF analysis, cells were collected, washed, resuspended in media and stained for viability with Cell-ID Intercalator-103Rh for 15 min at 37°C. To avoid non-specific staining, cells were subsequently blocked in flow buffer supplemented with either anti-mouse CD16/CD32 antibody (eBioscience) or Human TruStain FcX Fc Receptor Blocking Solution (BioLegend) for 30 min on ice. For phosphorylation experiments, THP1 cells were first labelled with a unique barcode by incubating with CD45-antibodies conjugated to distinct metal isotopes before pooling. Next, cells were stained for cell surface antigens, fixed and permeabilized using BD Cytofix/Cytoperm solution (BD Biosciences) and stained with the tag antibodies for 30 min on ice. For phosphorylation experiments, immediately after stimulation cells were incubated with 1% PFA on ice for 20 min, washed and fixed with pure methanol overnight in −80°C. After intracellular/tag staining, cells were washed and incubated in 125 nM Ir intercalator (Fluidigm) diluted in PBS containing 2.4% formaldehyde for 30 min at RT, washed and stored in PBS at 4°C. Immediately prior to acquisition, sampl es were washed once with PBS, once with de-ionized water and then resuspended at a concentration of 1 106 per ml in deionized water containing a 1:20 dilution of EQ 4 Element Beads (Fluidigm). The samples were acquired on either a CyTOF2 or Helios (both Fluidigm) equipped with a SuperSampler fluidics system (Victorian Airships) at an event rate of <500 events/second. After acquisition, the data were normalized using bead-based normalization using the CyTOF software. The data were gated to exclude residual normalization beads, debris, dead cells and doublets, leaving NGFR+ events for clustering and high dimensional analyses.
Western Blot
Rtp4 KO, Psmb8 KO or control sgRNA-transduced 4T1-Cas9-GFP cells were stimulated with 10 ng/ml IFNγ (Peprotech) for 48h. Western blot was performed according to the supplier’s instructions using rabbit monoclonal anti-Psmb8 antibody (Cell Signaling, clone D1K7X).
qPCR
Rtp4 KO, Psmb8 KO or control sgRNA-transduced 4T1-Cas9-GFP cells were stimulated with 10 ng/ml IFNγ (Peprotech) for 48h. RNA was extracted from cells using QIAzol Lysis Reagent (Qiagen) according to the manufacturer’s instruction. For cDNA synthesis, 1 μg total RNA was reverse-transcribed for 1 h at 37 °C with an RNA-to-cDNA kit (Applied Biosystems). For quantitative PCR, SYBR green qPCR master mix (Thermo Scientific) and the following primers were used: mouse Actb forward, 5’-CTAAGGCCAACCGTGAAAAG-3’, and reverse, 5-ACCAGAGGCATACAGGGACA-3’; mouse Rtp4 forward, 5-CGGGGCCAAGTGGAG-3’, and reverse, 5’-T GGCACAAGAT CATCACCT G-3’.
Sanger sequencing of the Rtp4 gene
To detect CRISPR/Cas9-induced gene editing of the Rtp4 gene, we isolated genomic DNA from cells using DNeasy Blood & Tissue Kilt (Qiagen). A 500 bp-size region flanking the target site of the Rtp4 gRNA (5’-ATCCAAATGCAGGCTCCACT-3’) was PCR amplified using DreamTaq polymerase (Thermo Fisher Scientific) and forward primer: 5’-TCTCTCCCAGATTTGAGGAAGA-3’, and reverse primer: 5’-AGCATGGGGACATGGAGTAC-3’. The PCR product was cloned into pCR®4-TOPO® plasmid using TOPO® TA Cloning Kit for Sequencing (Thermo Fisher Scientific) and transformed into TOP10 competent cells. Resulting colonies were then sequenced using M13 forward primer and aligned to the Rtp4 gene in the reference mouse genome.
Quantification and statistical analysis
Data visualization and analysis
CyTOF data was first debarcoded using Single Cell Debarcoder (Zunder et al., 2015) using post-assignment debarcode stringency filter and outlier trimming. Clean, concatenated files were then visualized using viSNE (Amir et al., 2013), a dimensionality reduction method, which uses the Barnes-Hut acceleration of the t-SNE algorithm. viSNE was implemented using either the Rtsne R package or Cytobank and generated using as input tag expression levels transformed by dividing by 5 and taking the arc-sine of the resulting value. Cell clusters were defined either by tag expression or in an unbiased way using the DBSCAN algorithm implementation in R after dimensionality reduction by t-SNE. Heatmaps of cell clusters were generated by taking the median untransformed or arc-sine transformed intensity within clusters and using this value unscaled or Z scaled.
Statistical analysis
All statistical details of experiments, including reproducibility (number of independent experiments performed), number of data point per group and definition of center and dispersion for each group are detailed in the figure legends. Heatmaps of cell clusters were generated by taking the median untransformed or arc-sine transformed intensity or the percentage of negative cells within clusters and using this value unscaled or Z scaled relative to other cell clusters.
Additional Resources
An extensive user guide for step-by-step cloning a Pro-Code/CRISPR library can be found in Supplementary information.
Supplementary Material
HighlightsTABLE.
We generated 100s of genetic barcodes detectable as proteins (Pro-Codes)
Pro-Codes provide a single-cell resolution means for vector and cell tracking
Pro-Codes enable high-dimensional phenotyping for CRISPR screens
This approach identified roles for Rtp4, Psmb8 and Socs1 in cancer immune editing
ACKNOWLEDGMENTS
We thank J. Brody and L. Devi (Mount Sinai), J. Agudo (Harvard), and A. Telesnitsky (Michigan) for helpful discussions, A. Ventura (Sloan Kettering) and S. Elledge (Harvard) for helpful comments on the manuscript, and the Mount Sinai Human Immune Monitoring Center and Eduardo Farias for technical assistance. B.D.B. was supported by NIH R01AI113221 and R33CA182377. M.M. was supported by NIH U19AI128949. M.D. was supported by the Belgian American Educational Foundation. A.Baccarini was supported by the OCRFA Liz Tilberis Award. This work utilized a Helios mass cytometer supported by NIH S10OD023547.
Footnotes
DECLARATION OF INTERESTS
B. D.B. and A.W. have filed a patent application related to the Pro-Code technology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson B, Norman TM, Jost M, Cho MY, Nunez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson B, Norman TM, Jost M, and Weissman JS (2018). Approaches to maximize sgRNA-barcode coupling in Perturb-seq screens. BioRxiv 298349. [Google Scholar]
- Agudo J, Ruzo A, Park ES, Sweeney R, Kana V, Wu M, Zhao Y, Egli D, Merad M, and Brown BD (2015). GFP-specific CD8 T cells enable targeted cell depletion and visualization of T-cell interactions. Nat. Biotechnol. 33, 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agudo J, Park ES, Rose SA, Alibo E, Sweeney R, Dhainaut M, Kobayashi KS, Sachidanandam R, Baccarini A, Merad M, et al. (2018). Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 48, 271–285.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir ED, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, et al. (2014). Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarini A, Chauhan H, Gardner TJ, Jayaprakash AD, Sachidanandam R, and Brown BD (2011). Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr Biol 21, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M, Kirk CJ, and Groettrup M (2013). The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 25, 74–80. [DOI] [PubMed] [Google Scholar]
- Bassik MC, Lebbink RJ, Churchman LS, Ingolia NT, Patena W, LeProust EM, Schuldiner M, Weissman JS, and McManus MT (2009). Rapid creation and quantitative monitoring of high coverage shRNA libraries. Nat Methods 6, 443–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir E.-a. D, Krutzik PO, Finck R., Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. (2011). Single-Cell Mass Cytometry of Differential Immune and Drug Responses Across a Human Hematopoietic Continuum. Science (80-. ). 332, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini L a, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC,Kuchler A, Alpar D, and Bock C (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaillot FM, Rozenfeld R, Gupta A, and Devi LA (2008). Cell surface targeting of - opioid receptor heterodimers by RTP4. Proc. Natl. Acad. Sci. 105, 16045–16050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman D, Singh A, Garrity AJ, and Blainey PC (2018). Lentiviral co-packaging mitigates the effects of intermolecular recombination and multiple integrations in pooled genetic screens. BioRxiv 262121. [Google Scholar]
- Fread KI, Strickland WD, Nolan GP, and Zunder ER (2017). An unpdated debarcoding tool for Mass Cytometry with Cell Type-specific and Cell Sample-specific stringency adjustment. Pacific Symp. Biocomput. 22, 588–598. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-y Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesen C, Wang H. a O., Schapiro D, Zivanovic N, Jacobs A., Hattendorf B, Schüffler PJ, Grolimund D, Buhmann JM, Brandt S, et al. (2014). Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods. [DOI] [PubMed] [Google Scholar]
- Hill AJ, McFaline-Figueroa JL, Starita LM, Gasperini MJ, Matreyek KA, Packer J, Jackson D, Shendure J, and Trapnell C (2018). On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, and Amit I (2016). Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167, 1883–1896.e15. [DOI] [PubMed] [Google Scholar]
- King SR, Duggal NK, Ndongmo CB, Pacut C, and Telesnitsky A (2008). Pseudodiploid Genome Organization Aids Full-Length Human Immunodeficiency Virus Type 1 dNa Synthesis. J. Virol. 82, 2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Song IH, Heo S-H, Kim Y-A, Park IA, Bang WS, Park HS, Gong G, and Lee HJ (2018). Expression of Immunoproteasome Subunit LMP7 in Breast Cancer and Its Association with Immune-Related Markers. Cancer Res. Treat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Neff NF, Quake SR, and Weissman IL (2011). Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 29, 928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, and Wherry EJ (2016). Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 165, 272–275. [DOI] [PubMed] [Google Scholar]
- Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, and Brown BD (2012). High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods 9, 840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma A, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science (80-. ). eaao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. (2017). Identification of essential genes for cancer immunotherapy. Nature 548, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, Moore R, McClanahan TK, Sadekova S, and Klappenbach JA (2017). Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939. [DOI] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Xu Q, Li MZ, and Elledge SJ (2016). Sources of Error in Mammalian Genetic Screens. G3: Genes|Genomes|Genetics 6, 2781–-2790.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, and Hacohen N (2016). Neoantigens encoded in the cancer genome. Curr. Opin. Immunol. 41, 98–103. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, and Zhang F (2015). High-throughput functional genomics using CRISPR-Cas9.Nat. Rev. Genet. 16, 299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova DM, and Verkhusha VV (2013). Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods 10, 751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunder ER, Finck R, Behbehani GK, Amir E-AD, Krishnaswamy S, Gonzalez VD, Lorang CG, Bjornson Z, Spitzer MH, Bodenmiller B, et al. (2015). Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 10, 316–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.