
Reorganization of the host epigenome by a viral oncogene
- Roberto Ferrari1,2,
- Trent Su1,3,
- Bing Li1,
- Giancarlo Bonora1,4,
- Amit Oberai1,
- Yvonne Chan1,
- Rajkumar Sasidharan5,
- Arnold J. Berk6,7,
- Matteo Pellegrini2,5 and
- Siavash K. Kurdistani1,2,7,8,9
- 1Department of Biological Chemistry, David Geffen School of Medicine, University of California, Los Angeles, California 90095, USA;
- 2Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research, University of California, Los Angeles, California 90095, USA;
- 3Division of Oral Biology and Medicine, School of Dentistry, David Geffen School of Medicine, University of California, Los Angeles, California 90095, USA;
- 4UCLA Bioinformatics Interdepartmental Degree Program, David Geffen School of Medicine, University of California, Los Angeles, California 90095, USA;
- 5Department of Molecular, Cellular, and Developmental Biology, University of California, Los Angeles, California 90095, USA;
- 6Department of Microbiology, Immunology and Molecular Genetics, University of California, Los Angeles, California 90095, USA;
- 7Molecular Biology Institute, University of California, Los Angeles, California 90095, USA;
- 8Department of Pathology and Laboratory of Medicine, David Geffen School of Medicine, University of California, Los Angeles, California 90095, USA
Abstract
Adenovirus small e1a oncoprotein causes ∼70% reduction in cellular levels of histone H3 lysine 18 acetylation (H3K18ac). It is unclear, however, where this dramatic reduction occurs genome-wide. ChIP-sequencing revealed that by 24 h after expression, e1a erases 95% of H3K18ac peaks in normal, contact-inhibited fibroblasts and replaces them with one-third as many at new genomic locations. The H3K18ac peaks at promoters and intergenic regions of genes with fibroblast-related functions are eliminated after infection, and new H3K18ac peaks are established at promoters of highly induced genes that regulate cell cycling and at new putative enhancers. Strikingly, the regions bound by the retinoblastoma family of proteins in contact-inhibited fibroblasts gain new peaks of H3K18ac in the e1a-expressing cells, including 55% of RB1-bound loci. In contrast, over half of H3K9ac peaks are similarly distributed before and after infection, independently of RB1. The strategic redistribution of H3K18ac by e1a highlights the importance of this modification for transcriptional activation and cellular transformation as well as functional differences between the RB-family member proteins.
Adenovirus 5 (Ad5), a DNA tumor virus, encodes a highly conserved 243-amino acid protein named small E1A protein (e1a) that drives quiescent mammalian cells into S-phase by overcoming cellular processes that normally inhibit inappropriate cell cycling (Berk 2005). The ability of e1a to transform cells depends on its interactions with several host cell proteins, particularly the retinoblastoma (RB)-family proteins (RB1, RBL1 [p107] and RBL2 [p130]) and the closely related EP300 and CREBBP lysine acetyltransferases (KATs) (Sherr and McCormick 2002; Berk 2005; Ferrari et al. 2008; Horwitz et al. 2008). We showed previously that during the initial 24 h of infection, e1a associates with a large fraction of human gene promoters in a temporally ordered manner, resulting in redistribution of host transcription cofactors such as RB1 and EP300 and reprogramming of gene expression for cell replication (Ferrari et al. 2008). The interaction of e1a with EP300/CREBBP KATs (Horwitz et al. 2008), the major enzymes responsible for H3K18ac in vivo, causes ∼70% reduction in total cellular levels of H3K18ac but not of several other histone modifications (Horwitz et al. 2008; Jin et al. 2011). The dynamic binding of e1a to the genome was accompanied by relative reduction of H3K18ac at most gene promoters but increased levels at promoters of genes involved in cell cycling and DNA replication (Ferrari et al. 2008). The previous data were obtained using chromatin immunoprecipitation combined with DNA microarrays (ChIP-chip) that contained probes covering primarily gene promoters. Since promoter regions represent a small fraction of the genome, the ChIP-chip experiments did not provide a molecular explanation for the global decrease in the levels of H3K18ac or how it differs from H3K9ac, another histone acetylation site that is also associated with gene expression.
In this study, we used ChIP coupled with massive parallel sequencing (ChIP-seq) (Johnson et al. 2007) to determine the genome-wide distributions of H3K18ac and H3K9ac in human primary lung fibroblasts (IMR90) before and after infection with the Ad5 mutant dl1500 which expresses e1a with little or no expression of other viral genes (Montell et al. 1982). We found that 95.4% of peaks of H3K18ac that exist prior to expression of e1a were deacetylated by 24 h after infection, essentially erasing the normal pattern of H3K18ac. New peaks of H3K18ac are established primarily at the promoter regions of highly induced genes that function in cell replication. Approximately 25% of these new peaks are found in regions that were bound by RB1 and to some extent by RBL2 and RBL1 prior to e1a expression. Altogether, the e1a-expressing cells have 33% as many peaks of H3K18ac as in uninfected cells, accounting for the global decrease in H3K18ac. In contrast, the total number of peaks of H3K9ac increased by 28.2% after infection. Interestingly, we also found distinct peaks of nucleosomes acetylated on H3K9 and H3K18 associated with the viral genome.
Results
General experimental approach
IMR90 fibroblasts were grown to confluence and cultured an additional day before they were either mock-infected or infected with dl1500. Twenty-four h post-infection (p.i.), cells were harvested for mRNA-seq and ChIP-seq analyses. For all ChIP-seq experiments, the input DNA was also sequenced. Sequenced reads were aligned to the human genome (Hg19), and only those that matched a unique location with up to two sequence mismatches were retained (Supplemental Table S1). To define peaks of enrichment, we segmented the human genome into 100-bp windows and compared the ChIP and normalized input DNA read counts in each window. Using the Poisson distribution, we calculated P-values (P-val) for the enrichment of ChIP reads in each window. A cut-off P-val was determined for each experiment to maintain a false discovery rate (FDR) ≤ 1.5%. For histone acetylation and RB-family ChIPs, significant windows were defined as those with a P-val < 10−3 and P-val < 10−4, respectively, and with significant peaks at the same P-vals in the two neighboring windows (see Supplemental Methods). To detect H3K18ac, we raised a polyclonal antibody (814) that showed specificity, not only in ELISA and peptide dot blots but also in ChIP experiments, with yeast strains harboring mutations in histone H3 lysines (Suka et al. 2001; Supplemental Fig. S1A,B). As expected, cells expressing e1a showed a significant decrease in global H3K18ac levels when using the 814 antibody (Supplemental Fig. S1C). All analyses of H3K18ac enrichment were done in parallel with H3K9ac, since this mark is also associated with transcriptional activation, but a global change in its level in response to e1a expression was not observed (Horwitz et al. 2008; Supplemental Fig. S2A).
ChIP-seq reveals genome-wide hypoacetylation of H3K18 caused by adenovirus e1a
To determine how the distributions of H3K18ac and H3K9ac were affected by e1a expression, we performed ChIP-seq from mock- and dl1500-infected cells 24 h p.i. We found 16,660 peaks of H3K18ac in mock- and 5500 in dl1500-infected cells, with only 772 peaks shared between the two conditions, indicating that 95.4% of the H3K18ac peaks in mock-infected cells were absent in the infected cells (Fig. 1A). In the e1a-expressing cells, the total number of H3K18ac peaks was reduced to 33% of the number in mock-infected cells. The number of H3K9ac peaks in infected cells increased by 28.2% compared to the number in mock-infected cells, with 53% of the peaks in mock-infected cells also present in the dl1500-infected cells (Fig. 1B). When we compared the peak coverage area (i.e., number of kb covered by all peaks) for H3K18ac and H3K9ac, we found 76.6% lower coverage by H3K18ac but 12% more coverage by H3K9ac in the infected cells (Supplemental Table S1). Thus, the ChIP-seq of H3K18ac in dl1500- versus mock-infected cells recapitulated the observed loss of H3K18ac observed by immunofluorescence, Western blotting, and protein mass spectrometry (Horwitz et al. 2008). H3K18ac reduction by e1a is not simply due to cell cycling, since cellular levels of H3K18ac were equivalent in a replicating asynchronous IMR90 cell population (Supplemental Fig. S2A), which has ∼35% of cells in S-phase compared to mock-infected contact-inhibited IMR90 cells with only 16% of cells in S-phase (Supplemental Fig. S2B).
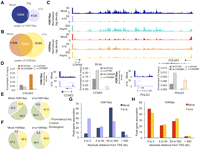
Small e1a causes global deacetylation and redistribution of H3K18ac. (A) Venn diagram showing the overlap between significant peaks of H3K18ac in mock- (dark blue) and dl1500-infected cells (light blue). (B) Venn diagram showing the overlap between significant peaks of H3K9ac in mock- (dark orange) and dl1500-infected cells (yellow). (C) Patterns of H3K18ac and H3K9ac in the intergenic region between COPS8 and COL6A3 genes in mock-, dl1500-infected, and asynchronous IMR90 cells. (D) Quantitative PCR (% of input) for EP300 and CREBBP in mock- and dl1500-infected cells for the COL6A3 intergenic region, and CCNE2 and POLD3 promoters are shown as bar plots. Patterns of H3K18ac in mock- and dl1500-infected cells at CCNE2 and POLD3 loci are also shown. For each histone modification, the y-axis indicates the number of input-normalized ChIP-seq reads across the locus (x-axis). (Light blue arrows) New peaks of acetylation in e1a-expressing cells. (Dark arrows) Direction of transcription. (E,F) Overview of distribution of H3K18ac and H3K9ac peaks in mock- and dl1500-infected cells in relation to gene structure are shown as pie charts. (G) Distribution of significant peaks of H3K18ac with respect to TSS in mock- (dark blue) and dl1500-infected (light blue) cells. (H) Distribution of significant peaks of H3K9ac with respect to TSS in mock- (dark orange) and dl1500-infected (yellow) cells.
Small e1a drastically alters the genomic distribution of H3K18ac
The global decrease of H3K18ac associated with e1a expression (Fig. 1A) was particularly evident at intergenic regions (IR), defined here as >3 kb upstream of the transcription start site (TSS) and >3 kb downstream from the transcription termination site (TTS) of all annotated genes. An example is the ∼200-kb locus on chromosome 2 between the COPS8 and COL6A3 genes where substantial levels of H3K18ac in mock-infected cells were essentially erased upon infection but maintained in asynchronously growing IMR90 cells (Fig. 1C; Supplemental Fig. S2C). The same IR retained similar levels of H3K9ac in all three conditions (Fig. 1C). ChIP-qPCR showed that loss of H3K18ac in e1a-expressing cells at the COPS8-COL6A3 locus was accompanied by a decrease in EP300 and CREBBP binding (Fig. 1D). Together with a substantial decrease in H3K18ac, we also observed the establishment of new peaks throughout the genome of e1a-expressing cells. Figure 1D shows two examples of cell cycle-regulated gene promoters, cyclin E2 (CCNE2) and DNA polymerase δ subunit 3 (POLD3), that gained H3K18ac in the infected cells. The gain of H3K18ac at these gene promoters was also confirmed by qPCR of ChIPed DNA (Fig. 1D; Supplemental Fig. S2D,E). ChIP-qPCR also revealed increased binding of EP300 to CCNE2 and of EP300 and CREBBP to the POLD3 promoters (Fig. 1D). Overall, we found that in mock-infected cells, H3K18ac was preferentially found in IRs (34%) and introns (54%), with only 12% of peaks at promoter regions (spanning 3 kb upstream of and downstream from the TSS) (Fig. 1E). The e1a-expressing cells showed a profoundly different distribution of H3K18ac, with IRs and promoter regions harboring 13% and 41% of the peaks, respectively (Fig. 1E). The H3K18ac peak distribution in introns was decreased to some extent in the infected compared to mock-infected cells (54% vs. 46%). The same analysis for H3K9ac showed that the distributions of the peaks in dl1500- vs. mock-infected cells were essentially unchanged (Fig. 1F). The redistribution of H3K18ac was also evident when we determined the absolute distance of significant peaks relative to the closest TSS. The majority of the H3K18ac peaks in mock-infected cells were positioned between 50 and 500 kb away from TSSs, with a small fraction within 5 kb (Fig. 1G, dark blue bars). In the dl1500-infected cells, however, the fraction of peaks mapping at regions closest to the TSS (5 kb) increased significantly, and the number of more distant peaks (50 to ≥500 kb) was decreased (Fig. 1G, light blue bars). The overall distribution of H3K9ac relative to the nearest TSS did not change substantially in the two conditions (Fig. 1H). We conclude that e1a induces a global decrease and considerable redistribution of H3K18ac by eliminating the majority of peaks in normal cells and establishing new, mostly promoter proximal peaks in the infected cells.
H3K18ac is increased at promoters of genes involved in cell cycling
To further characterize the changes in H3K18ac at promoters, we tiled the data from promoter regions of all genes from −5 to +5 kb of TSS into 100-bp windows. All promoters were grouped into three clusters that best captured the trend of the data based on the levels of H3K18ac in mock and infected cells using an unbiased clustering algorithm (k-means). The data are visualized as heat maps of enrichment values (Z-scores) in Figure 2A. The levels of H3K9ac for each promoter region are also shown. H3K18ac showed little correlation between mock and infected fibroblasts, while the H3K9ac pattern was more similar between the two conditions (Fig. 2A). The infected cells had 1154 promoters with at least one peak of H3K18ac (Fig. 2A, cluster 1), compared to 436 in mock cells (Fig. 2A, cluster 2). In contrast to H3K9ac, the peaks of H3K18ac in either condition were restricted to cluster 1 and 2 genes, indicating the specificity of H3K18ac distribution. The average change in expression (e1a-expressing vs. mock) of each cluster is shown in Figure 2B (see Methods).

Redistribution of H3K18ac at gene promoters is associated with gene expression changes in the e1a-expressing cells. (A) The distributions of H3K18ac and H3K9ac across ±5 kb from TSSs for all annotated transcripts in mock- and dl1500-infected cells are shown as heat maps. Each row represents one gene promoter. All promoters were grouped into three clusters based on k-means clustering of acetylation data. (B) Relative gene expression changes of the three clusters after e1a expression at 24 h p.i. Note the scale. (C,D) Patterns of H3K18ac and H3K9ac at representative genes from cluster 1 (CDT1 locus) and cluster 2 (CCL2 locus). (Light blue arrows) New peaks of H3K18ac in e1a-expressing cells. (Dark arrows) Direction of transcription. (E–G) GO analyses of genes from promoter clusters 1, 2, and 3 are shown, respectively. Bars represent −log10 of the P-value for the selected GO terms.
Cluster 1 genes showed little acetylation of H3K18 in mock-infected cells but a significant increase just downstream from the TSS in the infected cells, with little change in levels or distribution of H3K9ac (Fig. 2A). Gene ontology (GO) analysis of this cluster showed enrichment for genes involved in cell cycling, DNA replication, and ribosomes (Fig. 2E). Expression of this cluster was significantly up-regulated in the e1a-expressing cells (Z-score = 6.8, P < 0.0001), consistent with induction of S-phase and replication. Cluster 2 genes were associated with fibroblast function (Fig. 2F), which in mock-infected cells had high levels of H3K18ac both upstream of and downstream from the TSS. Upon infection, cluster 2 genes were significantly deacetylated on H3K18 and H3K9 (Fig. 2A) and repressed (Z-score = −20.6, P < 0.0001) (Fig. 2B). An example from each of cluster 1 and 2 genes is shown in Figure 2C,D, respectively. Cluster 3 promoters were enriched in membrane protein coding genes (Fig. 2G), without significant changes in acetylation (Fig. 2A), and showed marginal changes in gene expression (Z-score = 2.1, P = 0.011) (Fig. 2B). Taken together, our data indicate that e1a induces a general redistribution of H3K18ac from cell type-specific gene promoters, which are repressed, to those involved in cell growth and replication, which are induced.
The RB-family network in contact-inhibited IMR90 cells
Since a major requirement for induction of cell cycling by e1a is to overcome the RB1-mediated repression of cell cycle-regulated genes (Ghosh and Harter 2003; Berk 2005; Ferrari et al. 2008), we determined how the redistribution of H3K18ac induced by e1a relates to the binding of RB proteins before e1a expression. We performed ChIP-seq of all three RB-family members in contact-inhibited IMR90 cells. Our RB1 binding data in the contact-inhibited fibroblasts were very similar to a previous report on RB1 binding in quiescent IMR90 fibroblasts (Chicas et al. 2010; Supplemental Fig. S3). We found 1979, 1983, and 2336 significant peaks for RB1, RBL2, and RBL1, respectively (Fig. 3A). All three members were found commonly at gene promoters, with RB1 having the highest fraction of peaks in IRs (Fig. 3A). The average binding of each protein for all its target gene promoters, spanning ±3 kb of the closest TSSs, is shown in Figure 3B. Interestingly, while the peak of RBL2 is centered at the TSS, the peaks of RBL1 and RB binding are just upstream of and downstream from the TSS, respectively. Motif analysis using Galaxy SeqPos showed a differential enrichment for transcription factors (TFs) binding signatures in RB1 peaks compared to RBL2 and RBL1 (Fig. 3C). The RB1 target promoters were predominantly associated with E2F transcription factor motifs, whereas RBL2 and RBL1 target promoters contained motifs for other transcription factors (Fig. 3C).
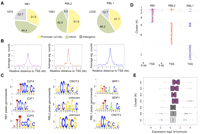
Analyses of RB-family protein genome-wide binding in contact-inhibited IMR90 fibroblasts. (A) Overview of RB1, RBL2, and RBL1 peak distributions in mock-infected cells in relation to gene structure are shown as pie charts. (B) Average binding profiles (significant counts) of the indicated RB-family members across the TSS regions of their respective target genes. (C) A 600-bp region around the peaks of RB1, RBL2, and RBL1 in mock-infected cells were analyzed for TF binding motifs using sitepro (Galaxy). Top three significant motifs for each family member are shown. All P-values are less than 1 × 10−10. (D) The distributions of the RB-family proteins across ±5 kb of TSS for genes with at least one RB-family member bound are shown as heat maps. The seven clusters are based on combinatorial binding patterns of the three proteins. (E) Relative gene expression changes of each of the seven clusters after e1a expression at 24 h p.i. are shown as box plots.
To determine whether the RB proteins have an overlapping set of target genes, we generated a tiling profile of their binding patterns across all promoters that had at least one of the RB-family proteins bound (Fig. 3D; Supplemental Fig. S4). We found 3942 genes with at least one RB-family member bound within ±5 kb of the TSS. These were grouped into seven clusters based on the combinatorial patterns of binding. Clusters 1–4 were bound by RB1, with clusters 1–3 bound by at least one of the RB1 paralogs. Clusters 5–7 were enriched for RBL1 and RBL2, RBL2 alone, and RBL1 alone, respectively (Fig. 3D; Supplemental Fig. S4). Notably, each of these clusters showed distinct, albeit overlapping, gene ontology enrichments (Supplemental Fig. S5), and their expression levels were differentially affected by e1a expression (Fig. 3E). Cluster 1, which contains 45 genes with all three RB-family proteins bound to their promoters, is highly enriched for DNA replication genes and was up-regulated by dl1500-infection more than any of the other clusters (Fig. 3E). Clusters 2–4, which are all bound by RB1 alone or with one other RB paralog, were also up-regulated after infection. However, clusters 5–7, which are RBL1 and RBL2 target genes, were less significantly induced as a group after infection (Fig. 3E). Altogether, the binding patterns of the RB-family proteins in uninfected cells are consistent with their known functions in control of the cell cycle and DNA replication, but each protein also has a unique set of target genes with differing expression changes in response to e1a expression.
Small e1a establishes new peaks of H3K18ac at and around regions bound by RB1 in uninfected cells
Since e1a removes the RB proteins from the promoters of cell cycle-regulated genes (Nevins et al. 1997; Ghosh and Harter 2003; Berk 2005; Ferrari et al. 2008; Sha et al. 2010), we next asked whether changes in H3K18ac or H3K9ac at promoter regions following e1a expression are related to the binding of the RB-family proteins prior to infection. We determined the significantly increased peaks of H3K18ac or H3K9ac in dl1500- vs. mock-infected cells and calculated the average change in the levels of acetylation in 100-bp intervals relative to the TSS for each of the clusters in Figure 3D. We found increased levels of H3K18ac in the promoter regions of genes in clusters 1–4, which were all bound by RB1 (Fig. 4A). Clusters 5–7 bound by RBL2 and RBL1 but without RB1 showed little to no change in the levels of H3K18ac relative to the average H3K18ac levels across the genome (i.e., the genome background) (Fig. 4B). None of the clusters showed any significant change in H3K9ac upon infection (Fig. 4C,D).
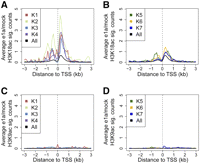
Relationship of the RB-binding clusters to histone acetylation change. (A,B) Average significant (sig.) counts of change in H3K18ac levels in dl1500- versus mock-infected cells are shown for the seven RB-clusters (see Fig. 3C). (C,D) Average significant (sig.) counts of change in H3K9ac levels in dl1500- versus mock-infected cells are shown for the seven RB-clusters (see Fig. 3C).
To explore the RB1 and H3K18ac relationship further, we selected and centered all the peaks of RB1, RBL2, and RBL1 throughout the genome in mock-infected cells. We then plotted the average levels of H3K18ac and H3K9ac in mock- and dl1500-infected cells within ±5 kb of each RB-family member peak center (Fig. 5A–F). We also calculated the fraction of H3K18 and H3K9 acetylation peaks in uninfected and e1a-expressing cells in the same interval (Fig. 5G–L). The levels of H3K18ac in mock-infected fibroblasts at RB1-bound target regions were minimal (Fig. 5A, purple line) but increased significantly in the infected cells (Fig. 5A, red line). The levels of H3K18ac also increased significantly at RBL2-bound target regions (Fig. 5B) and, to a minor extent, at RBL1-bound regions (Fig. 5C). The levels of H3K18ac in e1a-expressing cells also increased at RBL2-bound regions that were 5 kb away from RB1 peaks (Supplemental Fig. S6), indicating that new peaks of H3K18 were also established at RBL2-bound regions independent of RB1. Consistently, the number of new peaks of H3K18ac increased substantially at and around RB1-bound regions (Fig. 5G) as well as in regions bound by RBL2 (Fig. 5H) and less by RBL1 (Fig. 5I). Strikingly, 8% of H3K18ac peaks in the e1a-expressing cells occurred precisely where RB1 was bound, compared to 0.7% in mock-infected cells (Supplemental Fig. S7A). A similar but much smaller trend was observed for H3K18ac peaks that occurred precisely at RBL2- and RBL1-bound loci (Supplemental Fig. S7A). Interestingly, a double-peak of H3K18ac was observed only at RBL1-bound regions (cf. Fig. 5C and Fig. 5A,B). Such double-peaks of acetylation are commonly observed for acetylation marks relative to their HATs (Rada-Iglesias et al. 2011).
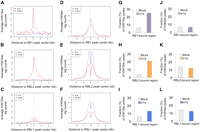
Genome-wide peaks of H3K18ac in the infected cells are established at regions that are bound by RB1, and to some extent RBL2, in uninfected cells. Average levels (sig. counts) of H3K18ac and H3K9ac in mock- and dl1500-infected (e1a) cells, respectively, are shown relative to (A,D) RB1-centered peaks, (B,E) RBL2-centered peaks, and (C,F) RBL1-centered peaks. (G–I) The fractions of H3K18ac peaks in mock- and dl1500-infected (e1a) cells that are within 5 kb of RB1, RBL2, and RBL1 peaks in mock-infected cells are shown as bar charts. (J–L) The same analysis as in G–I for H3K9ac.
When we examined the average H3K9ac levels at the RB protein peaks, we found a single peak of H3K9ac at the RB1-bound regions, with slightly lower levels of H3K9ac in the e1a-expressing cells (Fig. 5D). Remarkably, the H3K9ac profile showed a double-peak of acetylation around both the RBL2- and RBL1-bound regions in mock- and dl1500-infected cells, with lower levels in the latter (Fig. 5E,F). Consistently, the fraction of H3K9ac peaks colocalizing with any of the RB-family members did not change significantly between mock and e1a-expressing cells (Fig. 5J–L; Supplemental Fig. S7B). Taken together, these data suggest that a large fraction of the new H3K18ac peaks in the infected cells occur at or around regions that were bound by RB1 before e1a expression and, to a lesser extent, by RBL2 and RBL1. In addition, the different relationship between H3K18 and H3K9 acetylation and the RB-family members' binding sites suggests that H3K18 and H3K9 acetylation have different functions in gene regulation.
To gain further insight into the possible mechanism by which H3K18ac levels could be increased at clusters 1–4 (Fig. 4A), we analyzed published data sets of EP300 and CREBBP binding in the T98G glioblastoma line (Ramos et al. 2010), as well as EP300 and E2F1 binding in H9 human embryonic (hESCs) and MCF7 cells, respectively (Cao et al. 2011; Rada-Iglesias et al. 2011). In G1-arrested T98G cells, none of the seven RB-clusters showed significant enrichment for either EP300 or CREBBP (Supplemental Fig. S8A,C). However, upon serum stimulation, when cells are induced to enter S-phase as in e1a-expressing cells, clusters 1–4 showed greatly increased levels of EP300 binding compared to clusters 5–7 (Supplemental Fig. S8A–D). Upon serum stimulation, CREBBP showed increased binding to all clusters (Supplemental Fig. S8D). Also, in H9 hESCs, levels of EP300 binding were greater in clusters 1–4 compared to clusters 5–7 (Supplemental Fig. S8E). We also found that E2F1 binding in MCF7 cells is preferentially enriched in RB1-bound clusters 1–4 (Supplemental Fig. S8F). Finally, examination of e1a binding at a limited number of promoters showed that e1a may preferentially bind to RB1-bound gene promoters (POLD3 and CDC7) compared to those without RB1 (FOS and GAPDH) (Supplemental Fig. S9). Altogether, these data suggest that, compared to promoters bound by RBL2 or RBL1 without RB1, promoters bound by RB1 might have differential recruitment of EP300 and E2F1 as well as e1a itself. Such a scenario may also account for the preferential acetylation of H3K18 in clusters 1–4 versus 5–7 in the e1a-expressing cells.
We also found that 63% of intergenic RB1 peaks in uninfected cells were within 5 kb of H3K18ac peaks in e1a-expressing cells (Supplemental Fig. S10A). These RB1 peaks were enriched for TF binding motifs such as E4F1 and ATF2 (Supplemental Fig. S10B). Interestingly, E4F1 interacts with RB1, and E4F1 activity is regulated through the action of adenovirus e1a (Fajas et al. 2000). Surprisingly, the remaining IR RB1 peaks (390) that did not gain H3K18ac were enriched for E2F binding signatures (Supplemental Fig. S10C). These data suggest that e1a establishes new peaks of H3K18ac at a distinct subset of RB1-bound intergenic regions.
The new peaks of H3K18ac that are not within 5 kb of RB1 peaks were associated with 2533 genes and distributed between the TSS and mostly regions away from the TSS in e1a-expressing cells (Supplemental Fig. S11A). The majority of these genes were induced (Supplemental Fig. S11B) with significant enrichment for cell cycle genes, chromatin and RNA binding genes as well as TF motifs such as NFKB1, PLAGL1, TOPORS, and E2F4 (Supplemental Fig. S11C). The repressed genes were associated with fibroblast-related functions and TF motifs such as ZIC2, ZIC1, PLAG1, and AFFA4 (Supplemental Fig. S11D). These data suggest that e1a might exploit a distinct set of transcription factors or cofactors in addition to RB1 for acetylation of H3K18 and subsequent gene expression changes that favor the viral life cycle.
Specific intergenic regions are also targets of acetylation in infected cells
To characterize the H3K18ac redistribution at intergenic regions, we calculated the H3K18ac tag density per kb to determine a single enrichment value for H3K18ac at these locations (Fig. 6A). We found a small set of 406 IRs that gained new peaks of H3K18ac upon infection, compared to 1959 IRs that had drastically decreased levels of H3K18ac (Fig. 6A). The intergenic regions downstream from the ribosomal protein S6 kinase (RPS6KA1) and upstream of the Interleukin 6 (IL6) loci are examples of IRs where H3K18ac increased and decreased, respectively (Fig. 6B,C). Since H3K18ac at promoters is associated with active transcription, modulation of H3K18ac at IRs might also affect expression of the neighboring genes. To test this, we first performed gene ontology analysis of all the genes neighboring significant peaks of H3K18ac in IRs associated with mock- and dl1500-infected cells using the Genomic Region Annotation Tool (GREAT). We found that genes associated with H3K18ac in IRs of infected cells were enriched for genes activated by fibroblast growth factors and encoding transcriptional regulators (Supplemental Fig. S12A). In contrast, genes associated with IRs that decreased H3K18ac in infected cells are involved in differentiated fibroblast functions and immune response (Supplemental Fig. S12B). Genes associated with H3K18ac peaks in IRs of e1a-expressing cells were generally activated upon infection, while genes linked to IRs with decreased H3K18ac were generally repressed (Fig. 6D). Examples of genes associated with H3K18 acetylated IRs in the e1a-expressing cells include FOS and RPS6KA1, which are involved in regulation of cell proliferation, TAF1B, a basal transcription factor, and TFDP1, which heterodimerizes with E2F proteins to enhance their DNA-binding activity and promote transcription of genes involved in cell cycle progression (Polager and Ginsberg 2008; Fig. 6E). The genes associated with IRs that were deacetylated in the infected cells include IL1B, IRF1, IL6, and CCL2 which are all important mediators of the immune response (Akira and Kishimoto 1992; Feigenblum et al. 1998; Quinones et al. 2007). These results suggest that acetylation and deacetylation of H3K18 at IRs is also associated with induction and repression of transcription at a distance.

Intergenic peaks of H3K18ac coincide with potential distal regulatory elements to control gene expression. (A) The total H3K18ac tag density (number of sequence tags per kb) in intergenic regions (IRs) that are affected upon e1a expression is shown as a heat map. “Mock-IRs” and “e1a-IRs” are intergenic regions that harbor significant H3K18 acetylation in mock- or dl1500-infected cells, respectively. Distributions of H3K18ac, H3K9ac, and H3K4me1 in mock- and dl1500-infected cells around the RPS6KA1 (B) and the IL6 loci (C) are shown. (D) Relative gene expression changes of the e1a- and mock-IRs clusters after e1a expression at 24 h p.i. (E) Expression levels of candidate genes associated with e1a- and mock-IRs clusters are shown as heat maps. (F) Bar plot showing the fraction of H3K18ac peaks in the e1a-IRs that coincide with H3K4me1 in e1a-expressing cells.
To provide evidence that the H3K18 acetylated IRs in dl1500-infected cells may act as distal regulatory elements (Bulger and Groudine 2011), we performed ChIP-seq for H3K4me1, a histone methylation mark generally associated with enhancer elements (Heintzman et al. 2009; Hawkins et al. 2010; Rada-Iglesias et al. 2011) in e1a-expressing cells, and calculated the fraction of H3K18ac peaks in IRs that overlap with H3K4me1. We found that 65% of e1a-induced H3K18ac IR peaks occur preferentially at genomic locations that are also enriched for H3K4me1 (Fig. 6B,C,F). Analysis of potential TF binding motifs within 600 bp of peaks of H3K18ac in IRs of e1a-expressing cells yielded significant enrichment for TFs involved in cytokine-independent proliferation (e.g., SP1, PIR, GTF3C2, ERG1, and E2F1) (Zellmer et al. 2010; Supplemental Fig. S13A). The H3K18ac peaks in IRs of mock-infected cells were also highly enriched for TF binding signatures such as ZEB1, MZF1, REST, and NFKB1 (Supplemental Fig. S13B), all of which are involved in immune response and differentiation (Baeuerle and Henkel 1994; Hutton et al. 2004; Wang et al. 2009; Roizman 2011). These data suggest that the new H3K18ac peaks in IRs coincide with potential distal regulatory elements that promote cell growth and division, and the loss of H3K18ac from IRs may modulate the cell's response to viral infection and its phenotypic identity.
Histone acetylation marks specific regions of the Ad5 genome
Several reports have indicated that the adenovirus genome becomes associated with nucleosomes following infection (Sergeant et al. 1979; Karen and Hearing 2011; Komatsu et al. 2011; Ross et al. 2011). To determine if acetylated nucleosomes are localized to specific regions of the viral genome, we aligned the reads from input and acetylation ChIPs to the Ad5 genome (see Supplemental Methods) and searched for regions of enrichment using the same algorithm described above. We found several significant peaks of H3K9 and H3K18 acetylation across the viral genome (Supplemental Fig. S14). Our data are consistent with packaging of the Ad5 genome into chromatin, although the functional significance of histone acetylation relative to the hallmarks of the tightly packed viral genome remains to be determined.
Discussion
The interaction of adenovirus small e1a with the EP300/CREBBP KATs decreases the cellular level of H3K18ac in primary human cells to ∼30% of the level observed in uninfected cells (Horwitz et al. 2008). We now provide a molecular explanation for the global decrease in H3K18ac levels. Our data reveal that 95% of the H3K18ac peaks in uninfected cells are erased in the e1a-expressing cells, and one-third as many new peaks as before infection are established.
Loss of H3K18ac in the e1a-expressing cells was observed in both promoter and intergenic regions and was not a consequence of S-phase of the cell cycle per se. The affected promoter regions regulate genes with functions relevant to fibroblast biology and were repressed upon infection. Deacetylation of H3K18 in intergenic regions may also lead to deactivation of potential distant regulatory elements and to repression of associated genes with immune regulatory and fibroblast-related functions. This suggests that the e1a-induced transformation process involves suppressing the phenotypic identity of the infected cell and the cell's ability to mount a defense response by switching off the pertinent gene expression networks.
Small e1a establishes new peaks of H3K18ac at promoters of cell cycle-regulated genes that are highly induced and would eventually favor viral replication by driving the otherwise quiescent cell to proliferate. The new intergenic peaks of H3K18ac in dl1500-infected cells may also play a beneficial role for the virus by acting as enhancer elements for genes involved in cell proliferation. More intriguing is the fact that 55% of RB1-bound regions in the uninfected host cell gain significant levels of H3K18ac after infection. In fact, a subset of new H3K18ac occurs precisely where RB1 was bound prior to e1a expression. Although the genome-wide binding pattern of RB1 in the dl1500-infected cell is not known, numerous experimental results indicate that RB1 would be physically removed by e1a (Ghosh and Harter 2003; Berk 2005; Liu and Marmorstein 2007; Ferrari et al. 2008; DeCaprio 2009). So, the removal of RB1 by e1a is likely followed by acetylation of the underlying or surrounding nucleosomes on H3K18. This would be consistent with a model of e1a-targeted gene induction that not only requires removal of a transcriptional repressor (i.e., RB1) but also addition of an activating epigenetic mark (i.e., H3K18ac), probably through recruitment of EP300 and/or CREBBP (Ferrari et al. 2008; Horwitz et al. 2008). How H3K18ac effects are implemented by the cell is unknown, but e1a has highlighted a distinct function for H3K18ac among many histone acetylation sites including H3K9ac in regulating gene expression. Jin et al. (2011) also have recently reported that H3K18ac and H3K27ac, and not H3K9ac, are generated by EP300 and CREBBP and are required for activation of target genes by nuclear receptors.
The mechanism of genome-wide H3K18 deacetylation is not fully understood but requires interaction of e1a with EP300/CREBBP KATs (Horwitz et al. 2008), which are also relocalized upon infection (Ferrari et al. 2008). Whether one or more lysine deacetylases (KDACs), including certain sirtuins (SIRTs), which can deacetylate H3K18 in vitro (Black et al. 2008), are also involved remains to be determined. Interestingly, SIRT1 inhibition promotes differentiation (Pickard et al. 2010; Zhang et al. 2011), and its deletion induces inflammatory signaling in response to environmental stress (Schug et al. 2010; Zhang et al. 2010). This raises the possibility that H3K18 deacetylation could occur by sequestration of EP300/CREBBP and recruitment of these KATs to new but limited locations as well as active deacetylation by one or more KDACs to inhibit fibroblasts functions and antiviral responses and induce cell proliferation in the e1a-expressing cells.
Finally, our data have, for the first time, revealed the location of acetylated histones in the adenoviral genome. Together with previous reports that the adenoviral genome becomes chromatinized in the host cell (Sergeant et al. 1979; Karen and Hearing 2011; Komatsu et al. 2011; Ross et al. 2011), our data suggest that histone acetylation may also function to regulate the expression of adenovirus genes.
Methods
Cell culture and viruses
IMR90 human primary lung embryo fibroblasts (ATCC) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS) at 37°C in 5% CO2. Propagation of the dl1500 virus was done as described in Horwitz et al. (2008). Briefly, 293 spinner cells were grown in Joklik's modified Eagle minimal essential medium (SMEM) plus 5% FBS with 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine. Propagation of viruses was in 293 spinner cells with 2% FBS. Ad dl1500 stock titers were determined by plaque assay in duplicate on 293 cells.
Western blot
Isolation of core histones was performed as previously described (Pilch et al. 2004). The dried pellets were dissolved in 6× Laemmli sample loading buffer for subsequent analysis by Western blot. Western blots were performed using LI-COR.
Chromatin immunoprecipitation and library preparation
Chromatin immunoprecipitation for ChIP-seq was performed essentially as described (Ferrari et al. 2008) with few modifications. Briefly, 1 × 108 IMR90 lung fibroblasts were grown to 80% confluency (asynchronous population) or to confluence in 15-cm dishes. After 24 h, the cells were incubated with mock- or the dl1500- adenovirus for 1 h in media with 2% serum. After 24 h post-infection (p.i.), formaldehyde was added for 10 min at 37°C. After PBS washing, cross-linked cells were scraped from the plates and washed with 1 mL of PBS containing protease inhibitors (Roche). 2 × 107 cells were resuspended in 450 μl of lysis buffer and incubated for 10 min on ice and immediately sonicated using Misonix cup-horn sonicator. 100 μl of the lysate (corresponding to 5 × 106 cells) were used for each immunoprecipitation with a given antibody (listed below); 10 μl of the lysate were used as input. After overnight reversal of cross-linking at 65°C, samples were treated with RNase A for 30 min at 37°C and proteinase K for 2 h at 56°C. DNA was subsequently purified using phenol/chloroform extraction and precipitation. DNA concentration was measured using Qubit (Invitrogen). At least 10 ng of dsDNA for both input and IP were used for library preparation according to the manufacturer's instructions (Illumina). Libraries were sequenced using Illumina Genome Analyser II to obtain 76-bp-long reads (H3K9ac and corresponding input libraries) or Illumina HIseq-2000 to obtain 50-bp-long reads (H3K18ac and correspondent input libraries). ChIP for RB1 was performed as described (Chicas et al. 2010). Each library was checked for mismatch distribution.
A list of the antibodies used in this study is reported in the Supplemental Methods.
RNA extraction and mRNA-seq library preparation
Total RNA was extracted from mock- and dl1500-infected cells using the Qiagen easy RNA kit. The maximum amount of RNA was used to start the library preparation according to the manufacturer's instructions (Illumina). Libraries were sequenced using Illumina Genome Analyser II to obtain 76-bp-long reads. Alignment of mRNA-seq reads was performed using default parameters of TopHat (Trapnell et al. 2009; Kleindienst et al. 2010). Aligned reads were converted to sam format, and SAMMate software (Xu et al. 2011) was used to determine the transcript RPKM (reads per kilobase of exon per million of reads).
FACS analysis
FACS analysis has been carried out using the two-step EdU-PI staining kit (Life Technology) following the manufacturer's instructions.
Generation of H3K18ac antibody
A detailed description is given in the Supplemental Methods.
ChIP-qPCR
The same amount of chromatin was used for all ChIPs. ChIPed DNA was subjected to quantitative PCR using the respective input and IgG ChIP as control. qPCRs were performed using FastStart Universal SYBR Green Master (Rox). The ChIP final product (5 μl), 0.2 μM primers concentration, and 1× SYBR green mix were used for each reaction. qPCR analysis was performed using MxPro (Stratagene) with triplicate technical and biological experiments. ChIP-qPCR signals were calculated as a percentage of input. Standard deviations were measured and represented as error bars. Primers used for qPCR are listed in the Supplemental Methods.
ChIP-seq analysis
Reads were mapped to the Human (hg19) genome using Bowtie software. Only reads that aligned to a unique position in the genome with no more than two sequence mismatches were retained for further analysis. Duplicate reads that mapped to the same exact location in the genome were counted only once to reduce clonal amplification effects. The genome was tiled into 100-bp windows. Each read was extended by 150 bases (we refer to tags as the extend read counts within a bin) and was counted as one read to each window to which it partially or fully matched. The total counts of the input and ChIP samples were normalized to each other. The input sample was used to estimate the expected counts in a window; the average value for all windows was assigned to windows with zero counts. Finally, we used the Poisson distribution to estimate the probability of observing the ChIP counts within a window given the expected counts in the input sample window. We considered all windows with P-values less than 1.0 × 10−3 to have significant peaks. A P-value < 1.0 × 10−3 was chosen to give a False Discovery Rate (FDR) of <5%. The FDR was calculated by applying the same statistic described above to the two halves of the same input library. We considered the total number of significant peaks obtained this way as an estimate of the number of false-positive peaks. To compare two ChIP samples to identify regions that are significantly different between the two samples (for instance, e1a vs. mock), we replaced the input sample with the sample used for comparison; all downstream analyses were identical. Our algorithm produced several files that were subsequently used for analysis: BED files containing the coordinates of the significant windows of enrichment; Wiggle (wig) files (chromosome tiling, fixed step) with normalized read counts for the significant windows (if a window was not significant, we placed zero tags); GR files of normalized raw counts for input, and ChIP samples were also created for genome browser visualization (e.g., Fig, 1C,D). Tiling profiles of promoter regions for the hg19 annotated human promoters were also generated (Figs. 2A, 3D). These represent the 100-bp tiling of a 10-Kb region spanning the transcription start site. We reported the number of reads falling into significant windows and zero for the nonsignificant ones. Z-score scaling was applied to acetylation tiling profiles in Figure 2A in order to compare the levels of the different histone marks.
Detailed statistical methods are described in the Supplemental Methods.
Data access
All data in this work are available for download at the NCBI Gene Expression Omnibus (GEO) under accession number GSE32340.
Acknowledgments
G.B. was supported by a USPHS National Research Service Award, GM07104. Work in the A.J.B. lab is supported by R37CA025235. This work was supported by grants from the American Cancer Society and Howard Hughes Medical Institute and by an NIH Director's Innovator award to S.K.K.
Author contributions: R.F., A.B., and S.K.K. conceived and designed the experiments. R.F. performed experiments and data analysis. T.S. and Y.C. performed RNA-seq; T.S., B.L., G.B., A.O., R.S., and M.P. performed script programming and helped with data analysis and interpretation. A.B. provided dl1500 virus. R.F., A.B., and S.K.K. wrote the manuscript.
Footnotes
-
↵9 Corresponding author
E-mail skurdistani{at}mednet.ucla.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.132308.111.
- Received September 21, 2011.
- Accepted April 5, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








