




Abstract
Cutaneous T cell lymphoma is a heterogeneous group of lymphomas characterized by the accumulation of malignant T cells in the skin. The molecular and cellular etiology of this malignancy remains enigmatic and what role antigenic stimulation plays in the initiation and/or progression of the disease remains to be elucidated. Deep sequencing of the tumor genome revealed a highly heterogeneous landscape of genetic perturbations and transcriptome analysis of transformed T cells further highlighted the heterogeneity of this disease. Nonetheless, using data harvested from high-throughput transcriptional profiling allowed us to develop a reliable signature of this malignancy. Focusing on a key cytokine signaling pathway, previously implicated in CTCL pathogenesis, JAK/STAT signaling, we used conditional gene targeting to develop a fully penetrant small animal model of this disease that recapitulates many key features of mycosis fungoides, a common variant of CTCL. Using this mouse model, we demonstrate that T cell receptor engagement is critical for malignant transformation of the T lymphocytes and that progression of the disease is dependent on microbiota.
INTRODUCTION
Cutaneous T cell lymphoma (CTCL) is a heterogeneous group of non-Hodgkin’s lymphomas characterized by the accumulation of malignant T lymphocytes in the skin (Willemze 2005). CTCL patients typically present with erythematous, scaling skin patches, and plaques that can progress to tumors and widespread erythroderma (Hwang et al. 2008; Jawed et al. 2014). The most common variant of CTCL, mycosis fungoides (MF), is often indolent in its early stages and can be managed by topical agents or phototherapy. However, advanced stages of MF, and the leukemic variant of the disease, Sézary syndrome (SS), have a more aggressive clinical course, prove difficult to treat, are debilitating, and have no cure.
The age-adjusted incidence of CTCL is less than 10 cases per million people in the United States (Jawed et al. 2014). The rarity and heterogeneity of CTCL has made it difficult to understand the pathogenesis of this malignancy. While several studies have investigated the genetic changes in CTCL, there is little consensus as to the molecular drivers of this disease. Array-based comparative genome hybridization (CGH) and sequencing studies have demonstrated that CTCL is genetically unstable with various mutations as well as gains and losses of large parts of chromosomes (Vermeer et al. 2008; van Doorn et al. 2009; Choi et al. 2015). These aberrations have resulted in changes in cytokine signaling pathways and in the Rb, p53, and PTEN pathways (Vermeer et al. 2008; Wong et al. 2011; Lamprecht et al. 2012; Choi et al. 2015; da Silva Almeida et al. 2015; Kiel et al. 2015; McGirt et al. 2015; Wang et al. 2015). Changes in genes within cell survival pathways, the NF-κB pathway, and those involved with chromatin remodeling and DNA damage response have also been observed (Choi et al. 2015; Kiel et al. 2015). However, the contribution of the individual genetic perturbations to disease pathogenesis remains unclear.
The characteristic skin homing of malignant T cells in CTCL is notable, in part, because as a barrier surface, the epithelium is host to antigens that can trigger inflammatory responses contributing to malignant transformation. T cells in MF patients typically show characteristic signs of chronic antigenic stimulation (Girardi et al. 2004). Coupled with notable increases in antigen presenting cells in patient skin (Pigozzi et al. 2006), evidence indicated a role for T cell receptor signaling in the etiology of this disease. Several studies demonstrating the incidence of CTCL is higher among certain professions further highlight that exposure to specific environmental antigens may be a contributing factor (Morales-Suárez-Varela et al. 2005).
Commensal microbiota represent a primary source of antigenic exposure on the skin and a role for the skin-resident commensal Staphylococcus epidermis in tuning the inflammatory milieu within this tissue has been established (Naik et al. 2012). While S. epidermis promotes protective immunity at the skin barrier surface via enhancing Th17 differentiation, microbial dysbiosis has been shown to play a role in the initiation and progression of cancer in other contexts (Hu et al. 2013). In addition to commensal microbes, pathogenic infections have also been implicated in malignancy (Polk and Peek 2010; Elinav et al. 2013; Belkaid and Hand 2014). In line with this, CTCL patients frequently present with bacterial infections, particularly with Staphylococcus aureus, and antibiotic treatment to eliminate S. aureus often produces notable clinical improvements (Tokura et al. 1995; Jackow et al. 1997; Nguyen et al. 2008). While a link between microorganisms and CTCL initiation and/or progression has been noted (Axelrod et al. 1992; Jackow et al. 1997; Willerslev-Olsen et al. 2013), establishing a causative relationship between skin-resident and pathogenic bacteria and disease progression is nearly impossible in the absence of a reliable animal model of this malignancy.
In this study, whole exome sequencing (WES) of a cohort of SS patients revealed a heterogeneous spread of genetic alterations that converged on several oncogenic pathways, including PI3K signaling and STAT (Signal Transducer and Activator of Transcription) 3 pathway. Critically, gene set enrichment analysis (GSEA) of high throughput RNA sequencing data from malignant T cells demonstrated a distinct CTCL transcriptional signature validated with previously published transcriptome data. Using conditional gene targeting to express a hyperactive allele of STAT3 selectively in T lymphocytes, we generated an animal model of CTCL that recapitulates many of the key features of human disease. Generation of this model demonstrates the causative role of deregulated STAT3 signaling in CTCL pathogenesis. Further, using this model we also demonstrate that antigenic signaling and the presence of microbiota are necessary for CTCL progression, establishing a pre-clinical model for evaluation of therapeutic strategies for CTCL.
RESULTS
High throughput sequencing of malignant cells highlights genetic heterogeneity of SS
To gain insight into the molecular etiology of CTCL, we isolated T lymphocytes from the blood of SS patients. Transformed T lymphocytes were isolated at high purity based on the characteristic expression of CD7 and CD26. (Supplementary Fig. 1). Despite a large number of mutations found in malignant T cells, few genes were mutated in more than one patient, and no genes were mutated in three or more patients, emphasizing the molecular heterogeneity of this disease (Fig. 1a). In contrast, copy number variation (CNV) analysis of exomes revealed notable recurrent losses and gains of whole sections of chromosomes (Fig. 1b). Patients with a larger burden of circulating tumor cells tended to have more gross CNVs, indicating an escalation in chromosomal instability with disease progression. The most common chromosomal changes include the loss of 17p, gain of 17q, loss of parts of chromosome 10, and gain of 8q, with most of these changes previously observed (Fischer et al. 2004; Vermeer et al. 2008; Lamprecht et al. 2012; Cristofoletti et al. 2013; Kiel et al. 2015). Notable genes lost in 17p and chromosome 10 include the tumor suppressors TP53 and PTEN respectively, while gene copy numbers gained in 17q and 8q include STAT3 and STAT5, and MYC respectively, all well established to be associated with lymphomagenesis (Fig. 1b). Copy number amplifications of STAT3 and MYC, and loss of PTEN and TP53 were verified using digital droplet PCR (Supplementary Fig. 2). The observed diverse genetic aberrations converged on common cancer-associated signaling pathways in SS with many CNVs associated with the ERK/MAP-kinase, NFκB, PI3K-AKT, TP53, and STAT3 signaling pathways (Supplementary Fig. 3).
Fig. 1. Genetic landscape of CTCL.
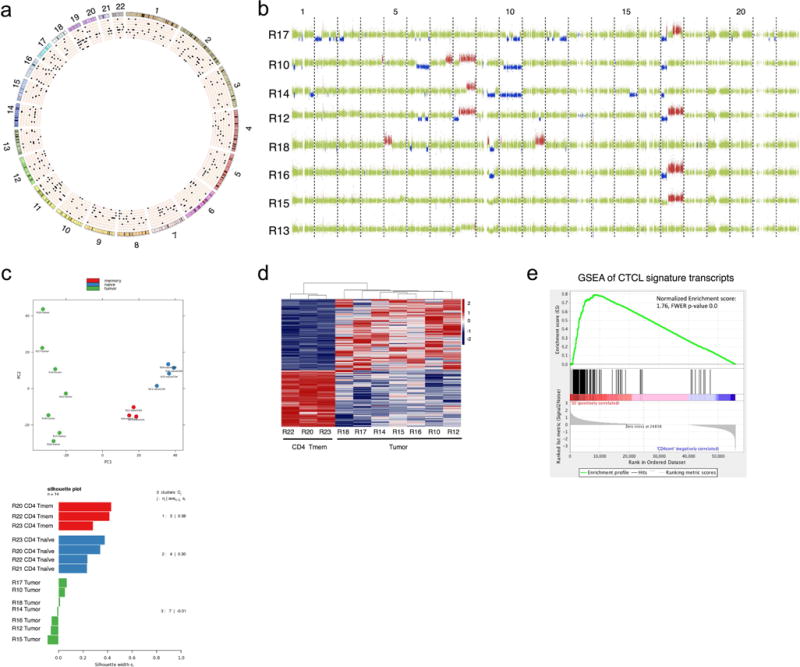
(a) Circos plot of SNVs identified by WES. Mutations were identified by comparing the tumor cells with patient’s B cells. Individual chromosomes are marked on the outer ring. Concentric circles represent patient genomes. Black pips indicate deleterious SNVs filtered for coverage >14 reads and predicted deleteriousness score of >0.5 by PolyPhen-2. 8 patient biospecimens are ordered from outside-in by decreasing tumor burden. (b) Copy number gains (red) and losses (blue) detected in SS genomes. Green indicates normal copy number. Each row represents analysis from an individual patient with samples ordered by decreasing tumor burden. (c) PCA plot (top) and silhouette analysis (bottom) of RNAseq data from 7 SS samples, and Tnaïve and Tmemory cells from healthy individuals. (d) Heat map displaying differential gene expression of malignant T cells (tumor) to memory T cells (memCD4), genes displayed have q value ≤ 0.05 (e) GSEA analysis performed on published transcriptome of sorted T cells from SS patients and healthy individuals using our CTCL gene signature.
Transcriptome analysis reveals a distinct CTCL gene expression signature
We next examined the gene expression profile of sorted T cells from patient tumor samples compared to naïve and memory T cells from healthy individuals. Principal Component Analysis (PCA) revealed that while the memory and naïve T cell populations from healthy patients clustered with their respective cell types, tumor samples were widely spread across the PCA plot highlighting the molecular heterogeneity of this disease (Fig. 1c). Silhouette analysis performed to measure the degree of similarity between clusters underscored the heterogeneity of the tumor cluster (Fig. 1c). Both MF and SS forms of CTCL are thought to arise from malignant transformation of memory T (Tmem) lymphocytes, and indeed this is consistent with our PCA analysis, as the malignant cells are closer to Tmem cells along PC1 than they are to naïve T lymphocytes. Analysis of differential gene expression between the transformed T cells and Tmem cells yielded a gene expression signature which we trimmed down to 124 genes upregulated in SS cells, based on q value (≤ 0.05) and fold difference in expression (>4×) (Fig. 1d, Supplemental Table 1). To validate the robustness of this signature we performed gene set enrichment analysis (GSEA) (Subramanian et al. 2005) on a previously published transcriptome of SS cells from 32 patients and sorted T lymphocytes from healthy individuals (Wang et al. 2015). Our CTCL signature was dramatically enriched in the malignant T cells from the published data set, despite the differences in cell isolation, library preparation and sequencing methodology (Fig. 1e, Supplemental Fig. 4), highlighting the potential of using this gene expression signature for diagnosis.
STAT3 inhibition results in decreased cell proliferation and survival in CTCL cell lines
While our analysis of the genetic landscape, along with other recently published whole exome sequencing studies (Choi et al. 2015; da Silva Almeida et al. 2015; Kiel et al. 2015; McGirt et al. 2015; Wang et al. 2015), have highlighted the molecular heterogeneity of CTCL, one pathway that is consistently upregulated in this disease is the STAT3 cytokine signaling pathway. Constitutive activation of STAT3 is an omnipresent feature of cell lines established from CTCL patients (Sommer et al. 2004; Krejsgaard et al. 2011; Netchiporouk et al. 2014). Many cytokines can trigger STAT3 phosphorylation and the subsequent activation of this signaling pathway contributes to the regulation of genes important in survival and proliferation. Our analysis of CNVs revealed that STAT3 duplications are observed in nearly half the patients (Fig 1b, Supplemental Fig. 2), with CNVs and single nucleotide variations (SNVs) also observed in phosphatases and kinases known to regulate STAT3 activity (Supplemental Fig. 3). These results are consistent with reports of cytogenetic amplifications of STAT3 from other genome-wide studies of CTCL (da Silva Almeida et al. 2015; Woollard et al. 2016). Two recent studies further emphasize the potential role of STAT3 in CTCL pathogenesis by identifying rare somatic mutations in the STAT3 SH2 domain, responsible for mediating dimerization, in malignant T lymphocytes from CTCL patients (da Silva Almeida et al. 2015; Kiel et al. 2015). To test the reliance of CTCL cells on STAT3 activity, we treated CTCL cell lines with STA-21, a selective STAT3 inhibitor (Song et al. 2005). The Sez4 cell line is derived from the blood of a SS patient (Abrams et al. 1991) and the MyLa 2059 line is derived from a plaque biopsy of an MF patient (Kaltoft et al. 1992). The two patient-derived cell lines that we tested showed decreased cell number and increased cell death following STAT3 inhibition (Fig. 2). These results are consistent with previous studies that have demonstrated apoptosis of CTCL cell lines following inhibition of STAT3 activity by siRNA or dominant negative form of STAT3 and together highlight the importance of this signaling pathway for survival of the malignant cells (Sommer et al. 2004; Verma et al. 2010; Krejsgaard et al. 2011).
Fig. 2. STAT3 regulates cell survival in CTCL.

Cultured Sez4 or MyLa cells were treated with 80μM STA-21 or DMSO. Total cell number and percentage of dead cells using trypan blue staining was determined at set time points. n=3 for all conditions. 3 independent experiments with multiple technical duplicates. 2way ANOVA with Sidak’s multiple comparison post-test. ** (p ≤ 0.01), *** (p≤ 0.001), **** (p≤ 0.0001). Values shown as mean ± SEM.
An autochthonous mouse model of CTCL pathogenesis demonstrates augmented Th17 differentiation and T cell transcriptional signature mirroring human CTCL
To further investigate the role of STAT3 in malignant transformation of T lymphocytes in which a hyperactive version of STAT3, STAT3C (Bromberg et al. 1999), is knocked into the Rosa26 locus with an upstream floxed stop cassette (R26STAT3Cstopfl). Excision of the stop cassette mediated by Cre recombinase leads to expression of a flag-tagged STAT3C and concomitant expression of EGFP from the IRES-GFP cassette (Casola et al. 2006). Analysis of thymocytes from young R26STAT3Cstopfl/+ CD4Cre mice and littermate controls revealed no notable differences in thymic T cell development (Supplemental Fig. 5). However, with age, R26STAT3Cstopfl/+ CD4Cre mice developed a lymphoproliferative-like disease with characteristic hair loss and scaly skin plaques (Fig. 3a). The skin phenotype of the mice progressed with age and was scored based on severity of disease. The mice on the lower end of the scale displayed dry skin and a range of hair loss, while on the more severe end of the scale animals developed obvious sores and lesions (see Methods for disease scale). By 8 months, a majority of the mice displayed a visible skin phenotype (Fig. 3b) with rare atypical lymphocytes with irregular, cerebriform nuclear appearance reminiscent of SS cells present in some blood smears from older mice (Supplemental Fig. 6).
Fig. 3. R26STAT3Cstopfl/+ CD4Cre mice develop a skin phenotype highly reminiscent of CTCL.

(a) Representative image of 6-month-old R26STAT3Cstopfl/+ CD4Cre and littermate control mice. (b) Phenotype progression in R26STAT3Cstopfl/+ CD4Cre mice. Darker shading indicates more severe phenotype. Scale ranges from (0) no phenotype to (5) moribund condition -see methods section for a full description. n = 132 mice. (c) Skin sections from ~10-month-old control and R26STAT3Cstopfl/+ CD4Cre mice, with a cluster of T cells, reminiscent of Pautrier microabscess in the knock-in animal. Scale bar = 50 μm. (d) Fold difference of CD3+CD4+ T cells isolated from skin of R26STAT3Cstopfl/+ CD4Cre and age matched control mice. Mean ± SEM from 30 independent experiments, n ≥ 32 for each genotype. P value was determined using a Wilcoxon Signed Rank Test. (e) Number of CD3+CD4+ T cells from peripheral lymph nodes of control and R26STAT3Cstopfl/+ CD4Cre mice. Mean ± SEM from 24 independent experiments, n ≥ 29 for each genotype. Significance assessed using the nonparametric two-tailed Mann-Whitney U test (f) (Left) Representative Ki67 staining of CD3+CD4+ T cells from peripheral lymph nodes of control and R26STAT3Cstopfl/+ CD4Cre mice. (Right) Quantification of Ki67+ CD3+CD4+ cells. Data is from 14 independent experiments. n≥16 for each genotype. For figures d–f significance values are as follows: ns (p > 0.05), * (p ≤ 0.05), ** (p ≤ 0.01), *** (p ≤ 0.001) **** (p ≤ 0.0001).
Immunofluorescent staining for CD3 in skin sections from older mice highlighted clustering of T cells reminiscent of Pautrier microabscesses at affected sites (Fig. 3c, Supplemental Fig. 7a,b), a pathognomonic clinical feature of CTCL (Jawed et al. 2014). Additionally acanthosis, hyperkeratosis, and parakeratosis reminiscent of CTCL (Shapiro and Pinto 1994) accompanied T cell accumulation and proliferation in the skin (Supplemental Fig. 7). Supporting these observations, flow cytometry of the skin showed a nearly 10-fold increase in CD4+ T cell number in R26STAT3Cstopfl/+ CD4Cre mice as compared to control animals (Fig. 3d). Deep sequencing of the T cell repertoire suggested that the disease remained polyclonal (Supplemental Fig. 8), consistent with early MF (Whittaker et al. 1991), as advanced CTCL often shows expansion of a clonal subpopulation (Kirsch et al. 2015).
Along with the augmented number of T cells in the skin, enlarged lymph nodes of R26STAT3Cstopfl/+ CD4Cre mice had dramatically increased numbers of CD4+ T cells (Fig. 3e). T cells isolated from secondary lymphoid organs of R26STAT3Cstopfl/+ CD4Cre mice exhibited an increase in activated/memory CD4+ T cells (CD44hi CD62Llo) (Supplemental Fig. 9a,b) and had a greater percentage of proliferating CD4+ T cells, positive for the Ki67 antigen (Fig. 3f). Flow cytometry analysis of cytokine expression in CD4+ T cells from the skin of R26STAT3Cstopfl/+ CD4Cre mice revealed a dramatic increase in IL-17 and IL-22 producing T cells compared to control animals (Fig. 4a,b). This is consistent with observations of increased IL-17A production from skin biopsies of MF patients (Cirée et al. 2004). This trend was also observed in T cells isolated from peripheral lymph nodes (Fig. 4a,b). The frequency of IFNγ producing cells in the skin was not significantly different, however we observed a lower frequency of IL-4 producing cells in the skin of mutant mice (Supplemental Fig. 9c,d). Consistent with the previously reported finding that STAT3 directly promoted RORγt transcription (Yang et al. 2008), we observed a greater number of RORγt+ IL-17− and RORγt+ IL-17+ cells in the skin and lymph nodes of R26STAT3Cstopfl/+ CD4Cre compared to littermate controls (Supplemental Fig. 9e,f). Indeed, upregulation of STAT3 in malignant T cells of CTCL patients may explain the high frequency of Th17 cells often observed in this malignancy (Krejsgaard et al. 2011; 2013) and may contribute to the inflammatory skin microenvironment in this disease.
Fig. 4. Expanded population of Th17 cells in R26STAT3Cstopfl/+ CD4Cre mice have a distinct CTCL transcriptional signature.

(a) Representative intracellular flow cytometry analysis of CD3+CD4+ T cells from the skin and peripheral lymph nodes of ~10-month-old R26STAT3Cstopfl/+ CD4Cre and control mice (b) Top: Quantification of cytokine production from CD3+CD4+ T cells isolated from skin R26STAT3Cstopfl/+ CD4Cre and control animals. 23 independent experiments. n≥25 for each genotype. Bottom: Th17 cytokine production in CD4+ T cells from peripheral lymph nodes. ≥25 independent experiments. n≥28 for each genotype. Statistical significance was assessed using the nonparametric two-tailed Mann-Whitney U test. Significance values are as follows: ns (p > 0.05), * (p ≤ 0.05), ** (p ≤ 0.01), *** (p ≤ 0.001) **** (p ≤ 0.0001). Values shown as mean ± SEM (c) GSEA comparing the transcriptional profile of T cells from the skin of R26STAT3Cstopfl/+ CD4Cre mice vs controls using the established human CTCL gene signature.
To further assess the relevancy of our mouse model to human disease we examined the transcriptional profile of CD4+ T cells isolated from the skin of R26STAT3Cstopfl/+ CD4Cre mice. As shown in Fig. 4c, GSEA reflected that malignant T cells from the skin of R26STAT3Cstopfl/+ CD4Cre mice had a distinct, altered transcriptional pattern compared to CD4+ T cells sorted from the skin of control animals, and that our previously characterized (Supplemental Table 1) human CTCL gene expression signature was readily identifiable in the lymphocytes from mutant mice.
Disease progression in a CTCL mouse model is dependent on TCR signaling and the presence of microbiota
Cutaneous lymphomas are unique in that the malignant cells localize to surfaces, where environmental exposure in the form of pathogens or irritants may contribute to CTCL pathogenesis. Previous epidemiological studies have noted geographical clustering of this malignancy (Moreau et al. 2014; Litvinov et al. 2015; Ghazawi et al. 2017) suggesting an environmental trigger in disease initiation and a number of studies have implicated microbial contribution to CTCL initiation and progression (Axelrod et al. 1992; Tokura et al. 1995; Jackow et al. 1997). In particular, S.aureus is reported to be found on affected skin of 44-63% of CTCL patients across two prior studies (Nguyen et al. 2008; Talpur et al. 2008). Persistent activation of T cells via the antigen receptor by bacterial antigens and/or super-antigens may contribute to CTCL pathogenesis. To examine if T cell receptor (TCR) engagement is critical for CTCL pathogenesis we crossed the R26STAT3Cstopfl/+ CD4Cre mice onto an OTII Rag2 knock-out background to restrict the TCR repertoire. The OTII transgene encodes the TCRα and TCRβ chains of the T cell antigen receptor specific for the chicken ovalbumin peptide and the absence of the Rag2 enzyme ensures that no other TCR specificities are present in these animals. Analysis of R26STAT3Cstopfl/+ CD4Cre OTII Rag2KO mice revealed no expansion of T cells in the skin and, in contrast to older R26STAT3Cstopfl/+ CD4Cre littermates, these animals failed to develop any signs of clinical disease (Fig. 5a).
Fig. 5. Disease symptoms of R26STAT3Cstopfl/+ CD4Cre mice are ameliorated in TCR limited and germ free settings.

(a) Average phenotype score of R26STAT3Cstopfl/+CD4Cre (blue line), R26STAT3Cstopfl/+ CD4Cre Rag2KO OTII (orange line), and control mice (black line). Scale ranges from (0) no phenotype to (5) moribund condition -see methods section for a full description. Results are mean ± SEM (b) Average phenotype score of R26STAT3Cstopfl/+ CD4Cre and control animals housed under Specific Pathogen Free (SPF) or Germ Free (GF) conditions. Results are mean ± SEM (c) Evaluation of pruritus over time normalized to average of control mice at each time point. n=3 per genotype aged <5 months, n= 5 mice for each genotype above 5 months. See methods for details of video monitoring protocol. 2 way ANOVA with Bonferroni post-test. * (p ≤ 0.05), **** (p≤ 0.0001).
Given the often-noted bias in TCR Vβ repertoire in patient biospecimens (Linnemann et al. 2004), and the vast clinical experience with CTCL that suggests that worsening of symptoms is often associated with bacterial sepsis, with improvement of disease parameters following antibiotic therapy (Tokura et al. 1995; Talpur et al. 2008), we sought to examine the contribution of microbiota to disease initiation and progression in the R26STAT3Cstopfl/+ CD4Cre model. We rederived our CTCL mouse model into germ free (GF) isolators via hysterectomy and cross fostering to generate mice that we confirmed to be free of bacteria, viruses, and fungi via culture, qPCR and sequencing-based approaches. Remarkably, we observed that while the clinical signs of disease started at the same age in GF as conventionally housed animals (under specific pathogen free, SPF, conditions), the disease in GF never progressed to fulminant malignant disease (Fig. 5b) observed in the SPF animals. The course of disease was readily restored, once the R26STAT3Cstopfl/+ CD4Cre animals were cohoused with SPF animals (Fig. 5b).
The incendiary role of skin-resident bacteria and antigens they produce in CTCL pathogenesis is supported by the fact that S. aureus colonized the skin of CTCL patients at a higher rate than the general population (Talpur et al. 2008) and by the association between S. aureus sepsis or colonization and CTCL progression (Krejsgaard et al. 2014). S. aureus and other skin-associated opportunistic infections may contribute to disease progression through stimulation of T cells or via induction of cytokine production in by-stander cells, thereby contributing to the tumor microenvironment. We observed that much like MF patients, R26STAT3Cstopfl/+ CD4Cre animals present with notable pruritus (Ahern et al. 2012) (Fig. 5c), and the persistent scratching is likely to result in introduction of opportunistic infections. Given these observations, it is worth considering whether aggressive treatment of pruritus and prevention of S.aureus colonization in patients with early stage MF would reduce the chance of progression to more advanced CTCL disease.
DISCUSSION
Our genome- and transcriptome-wide analysis of sorted T cells from SS patients underscores the genetic heterogeneity of this malignancy. In agreement with a recent study that also highlighted a predominance of CNVs compared to SNVs in CTCL (Choi et al. 2015), our WES results suggest that CTCL may be a disease driven by CNVs instead of transformative point mutations. A few of the cytogenetic changes recurred in our cohort, including loss of 10q and 17p, and gain of 8q and 17q. Despite the diversity of individual mutations and cytogenetic alterations that the landscape analysis of CTCL revealed, the genetic perturbations consistently converged on the JAK/STAT pathway, along with the p53, MAPK, NFκB and PI3K pathways, in the malignant T cells. Changes to various genes within these pathways have previously been observed by whole exome sequencing and CGH studies, nonetheless there is a lack of consensus regarding the specific molecular drivers of CTCL (Vermeer et al. 2008; de Leval et al. 2009; Choi et al. 2015). We posit that rather than a single genetic driver of malignant transformation, the collective SNVs and CNVs alter several pathways relevant to lymphomagenesis. Since the majority of these mutational changes were copy number aberrations, the cumulative chromosomal instability may be a key mechanism of transformation in this disease.
While constitutive phosphorylation of STAT3 and the key role of STAT3 in the survival of CTCL cell lines has previously been documented (Sommer et al. 2004; Krejsgaard et al. 2011; McKenzie et al. 2011), our results help elucidate the molecular basis for the persistent STAT3 activation observed in CTCL and establish that this pathway is a driver of this malignancy. We found STAT3 was duplicated in three out of eight patients, but we also observed amplification of several kinases known to phosphorylate STAT3, such as JAK3 and SRC-family kinases (Bromberg and Darnell 2000), as well as loss of some of the negative regulators of STAT3. Our analysis of the heterogeneous genetic landscape and transcriptome of this enigmatic malignant disease yielded a defining transcriptional signature of CTCL. This presents a compelling potential diagnostic tool that warrants further investigation.
Using conditional gene targeting we demonstrated the critical role of cytokine signaling, namely STAT3, in promoting CTCL pathogenesis and established a tractable and spontaneous model of CTCL. Previously published mouse models of CTCL relied on xenograft transplantation of cell lines established from human CTCL patients or on injection of virally transduced mouse cells into immunocompromised mice (Charley et al. 1990; Thaler et al. 2004; Krejsgaard et al. 2010; Han et al. 2012; Ito et al. 2014; Wu et al. 2014; Adachi et al. 2015; Kittipongdaja et al. 2015), while another implanted a murine T cell lymphoma line into a syngeneic host to generate malignant T cell disease following DNFB stimulation (Wu et al. 2011). It is important to note, that while our genomic landscape analysis that highlighted the important role of STAT3 in human malignancy focused on genomic material from SS patients, our small animal model more closely mimicked the MF form of CTCL. Furthermore, our genetic approach of expressing the hyperactive mutant of STAT3 in all T lymphocytes, precludes us from making any inference regarding the precise cell of origin for this malignant disease – a topic that is hotly debated in the field (Campbell et al. 2010; Krejsgaard et al. 2017). Nonetheless, the relevant genetic trigger, the characteristic histopathological presentation of the disease, and the presence of a transcriptional signature of human malignancy in T cells from the mutant animals, all distinguish the autochthonous mouse model generated here. Further, because this model does not rely on injection or grafting of already malignantly transformed tissue, the development and progression of CTCL, along with genetic and pharmacological interventions, can be assessed.
Pathogenesis in this model is linked to the expression of pro-inflammatory Th17 cytokines. This is intriguing in light of the normally characterized clinical progression of CTCL. Skin lesions from patients in early stages of disease show enhanced expression of Th1 cytokines, such as IFNγ, thought to be linked to an anti-tumor immune response (Bagot et al. 1998; Echchakir et al. 2000; Kim et al. 2005). As the disease progresses the tumor microenvironment takes on a markedly different profile, characterized by an increase in Th2 cytokines, such as IL-4, IL-5, and IL-13, accompanied by a reduction in anti-tumor Th1 cells (Krejsgaard et al. 2017). However, several studies find enhanced expression of the cytokines IL-17A and IL-17F in lesional skin driven by STAT3 hyperactivity (Cirée et al. 2004; Krejsgaard et al. 2011; 2013). Enhanced expression of IL-17 is associated with an aggressive disease course in the subset of patients where it is seen (Krejsgaard et al. 2013; 2017). Our observation that mice expressing the hyperactive STAT3 allele display a Th17 biased inflammatory environment is consistent with the critical role that STAT3 plays in Th17 differentiation and thus the model may represent the more aggressive form of the disease.
Additionally, we took advantage of the CTCL animal model to demonstrate the central role of T cell receptor signaling in the development of CTCL. This will facilitate further explorations of the role of T cell interaction with antigen presenting cells in the development of the disease. Further, we confirm the necessity for microbial triggers in CTCL disease progression, thus validating the many epidemiological and clinical observations that have previously linked CTCL pathogenesis and bacterial infections. Use of this model can facilitate exploration of interventions that modify the composition of the skin microbiome as potential therapies. Given the molecular heterogeneity of this malignancy, we believe that targeting of the tumor microenvironment has to be considered along with inhibition of specific signaling networks found to be aberrantly expressed in the individual tumors. These interventions would have to truly reflect personalized medicine, with skin microbiome analysis complementing tumor RNA and DNA sequencing.
MATERIALS & METHODS
For detailed Materials and Methods, please see the Supplemental Materials
Clinical samples
Eight patients with SS and four healthy volunteers were identified at NYU Langone Medical Center and included in this study in accordance with protocols approved by the NYU School of Medicine Institutional Review Board and Bellevue Facility Research Review Committee (ClinicalTrials.gov ID: NCT01663571). CTCL patients were diagnosed according to the WHO classification criteria. Patients with history of other hematopoietic malignancies were not included in this study. After written informed consent was obtained, peripheral blood samples were harvested.
Quantification of disease progression in mouse model
Monthly phenotype scoring was performed in a blinded fashion. Skin phenotype was assessed and mice assigned a score of 0 to 5. Score of 0: wild type appearance; Score of 1: hair loss around eyes; Score of 2: dry skin, obvious scratching, thinning of hair on neck; Score of 3: extensive hair loss on face or back of neck; Score of 4: large bald or scaly patches of skin; Score of 5: Large scabs, sores, or open lesions. Mice with a score of 5 were euthanized and the score of 5 was carried throughout the remainder of the analysis.
Supplementary Material
Acknowledgments
We thank Drs. Markus Schober and Cindy Loomis for technical advice and for thoughtful discussions on skin biology. We thank Dr. Klaus Rajewsky for his thoughtful insight. We also thank the following NYU SoM Core Facilities for expert assistance: Histopathology, Immunohistochemistry, Flow Cytometry, Biorepository, and the Genome Technology Center. We thank Harini Babu from HistoWiz Inc, for her CD4 IHC work.
FINANCIAL SUPPORT
Work in the Koralov laboratory was supported by NIH R01HL125816, the NYUCI Pilot Grant, the Feinberg Lymphoma Research Grant, and grants from the Spatz Charitable Foundation, the Cutaneous Lymphoma Foundation, the Concern Foundation and Hirschl/Weill-Caulier Trust. Additionally, M.F. was supported by the NIH NIAMS Award Number F31AR070094. A.S. was supported by the William Randolph Hearst Foundation. A.S. and M.H.F. were supported by NIH Training Grants T32-GM007308, T32-CA009161, and T32-AI 100853-3. L.K.F. was supported by NIH F31 CA171596-02.The NYU Experimental Pathology Immunohistochemistry Core Laboratory is supported in part by the Laura and Isaac Perlmutter Cancer Center Support Grant; NIH/NCI P30CA016087 and the NIH S10 Grants; NIH/ORIP S10OD01058 and S10OD018338. N.Ø. was supported by the Novo Nordic Foundation Tandemprogram and the Danish Cancer Society Knaek Cancer Program. M.E and RSL were supported by R01 NIDCR (DE025639)
Abbreviations
- CTCL
Cutaneous T Cell Lymphoma
- MF
mycosis fungoides
- SS
Sézary syndrome
- CGH
comparative genome hybridization, WES: whole exome sequencing
- STAT
Signal Transducer and Activator of Transcription
- GSEA
gene set enrichment analysis
- CNV
copy number variation
- PCA
Principal Component Analysis
- Tmem
memory T cell
- SNV
single nucleotide variation
- TCR
T cell receptor
- GF
germ free
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors state no conflict of interest. Dr. Kutok is currently employed at Infinity Pharmaceuticals. His contribution to this work was prior to his employment there, while he was faculty at Brigham and Women’s Hospital.
AUTHOR CONTRIBUTIONS
MHF, AS, LKF, and SBK were directly involved in the design and execution of the experiments and writing of the manuscript. MHF performed all mouse experiments. AS, VN, SG, MEL, JL, and KBH were responsible for patient recruitment and biospecimen collection and processing. KK, ID, CL, and AH were responsible for sequencing and bioinformatics together with VN, AS, SBK. ME, RSL performed IF. MSS, JK, CL, NØ, KBH contributed to the interpretation of the results and direction of the project.
Online Supplemental material
Please see supplement for additional figures and methods.
References
- Abrams JT, Lessin S, Ghosh SK, Ju W. A clonal CD4-positive T-cell line established from the blood of a patient with Sézary syndrome. J Invest Dermatol. 1991;96(1):31–7. doi: 10.1111/1523-1747.ep12514693. [DOI] [PubMed] [Google Scholar]
- Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, et al. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med. 2015 Oct;19:1–10. doi: 10.1038/nm.3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern K, Gilmore ES, Poligone B. Pruritus in cutaneous T-cell lymphoma: a review. J Am Acad Dermatol. 2012 Oct;67(4):760–8. doi: 10.1016/j.jaad.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod PI, Lorber B, Vonderheid EC. Infections complicating mycosis fungoides and Sézary syndrome. JAMA. 1992 Mar 11;267(10):1354–8. [PubMed] [Google Scholar]
- Bagot M, Echchakir H, Mami-Chouaib F, Delfau-Larue MH, Charue D, Bernheim A, et al. Isolation of tumor-specific cytotoxic CD4+ and CD4+CD8dim+ T-cell clones infiltrating a cutaneous T-cell lymphoma. Blood. 1998 Jun 1;91(11):4331–41. [PubMed] [Google Scholar]
- Belkaid Y, Hand TW. Role of the Microbiota in Immunity and Inflammation. Cell. 2014 Mar 27;157(1):121–41. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg J, Darnell JE. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000 May 15;19(21):2468–73. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999 Aug 6;98(3):295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sézary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010 Aug 5;116(5):767–71. doi: 10.1182/blood-2009-11-251926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Segal J, et al. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci USA. 2006 May 9;103(19):7396–401. doi: 10.1073/pnas.0602353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charley MR, Tharp M, Locker J, Deng JS, Goslen JB, Mauro T, et al. Establishment of a human cutaneous T-cell lymphoma in C.B-17 SCID mice. J Invest Dermatol. 1990 Mar;94(3):381–4. doi: 10.1111/1523-1747.ep12874500. [DOI] [PubMed] [Google Scholar]
- Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015 Jul 20;47(9):1011–9. doi: 10.1038/ng.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirée A, Michel L, Camilleri-Bröet S, Jean Louis F, Oster M, Flageul B, et al. Expression and activity of IL-17 in cutaneous T-cell lymphomas (mycosis fungoides and Sézary syndrome) Int J Cancer. 2004 Oct 20;112(1):113–20. doi: 10.1002/ijc.20373. [DOI] [PubMed] [Google Scholar]
- Cristofoletti C, Picchio MC, Lazzeri C, Tocco V, Pagani E, Bresin A, et al. Comprehensive analysis of PTEN status in Sezary syndrome. Blood. 2013 Nov 14;122(20):3511–20. doi: 10.1182/blood-2013-06-510578. [DOI] [PubMed] [Google Scholar]
- da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat Genetics. 2015 Nov 9;47(12):1465–70. doi: 10.1038/ng.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leval L, Bisig B, Thielen C, Boniver J, Gaulard P. Molecular classification of T-cell lymphomas. Critical Reviews in Oncology/Hematology. 2009 Nov;72(2):125–43. doi: 10.1016/j.critrevonc.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Echchakir H, Bagot M, Dorothée G, Martinvalet D, Le Gouvello S, Boumsell L, et al. Cutaneous T cell lymphoma reactive CD4+ cytotoxic T lymphocyte clones display a Th1 cytokine profile and use a fas-independent pathway for specific tumor cell lysis. J Invest Dermatol. 2000 Jul;115(1):74–80. doi: 10.1046/j.1523-1747.2000.00995.x. [DOI] [PubMed] [Google Scholar]
- Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013 Nov 1;13(11):759–71. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- Fischer TC, Gellrich S, Muche JM, Sherev T, Audring H, Neitzel H, et al. Genomic aberrations and survival in cutaneous T cell lymphomas. J Invest Dermatol. 2004 Mar;122(3):579–86. doi: 10.1111/j.0022-202X.2004.22301.x. [DOI] [PubMed] [Google Scholar]
- Ghazawi FM, Netchiporouk E, Rahme E, Tsang M, Moreau L, Glassman S, et al. Comprehensive analysis of cutaneous T-cell lymphoma (CTCL) incidence and mortality in Canada reveals changing trends and geographic clustering for this malignancy. Cancer. 2017 May 10; doi: 10.1002/cncr.30758. [DOI] [PubMed] [Google Scholar]
- Girardi M, Heald PW, Wilson LD. The pathogenesis of mycosis fungoides. N Engl J Med. 2004 May 6;350(19):1978–88. doi: 10.1056/NEJMra032810. [DOI] [PubMed] [Google Scholar]
- Han T, Abdel-Motal UM, Chang D-K, Sui J, Muvaffak A, Campbell J, et al. Human Anti-CCR4 Minibody Gene Transfer for the Treatment of Cutaneous T-Cell Lymphoma. PLoS ONE. 2012 Sep 4;7(9):e44455–11. doi: 10.1371/journal.pone.0044455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Elinav E, Huber S. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci USA. 2013;110:9862–9867. doi: 10.1073/pnas.1307575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang ST, Janik JE, Jaffe ES, Wilson WH. Mycosis fungoides and Sézary syndrome. The Lancet. 2008 Mar;371(9616):945–57. doi: 10.1016/S0140-6736(08)60420-1. [DOI] [PubMed] [Google Scholar]
- Ito M, Teshima K, Ikeda S, Kitadate A, Watanabe A, Nara M, et al. MicroRNA-150 inhibits tumor invasion and metastasis by targeting the chemokine receptor CCR6, in advanced cutaneous T-cell lymphoma. Blood. 2014 Mar 6;123(10):1499–511. doi: 10.1182/blood-2013-09-527739. [DOI] [PubMed] [Google Scholar]
- Jackow CM, Cather JC, Hearne V, Asano AT, Musser JM, Duvic M. Association of erythrodermic cutaneous T-cell lymphoma, superantigen-positive Staphylococcus aureus, and oligoclonal T-cell receptor V beta gene expansion. Blood. 1997 Jan 1;89(1):32–40. [PubMed] [Google Scholar]
- Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. Diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014 Feb;70(2):205.e1–16. doi: 10.1016/j.jaad.2013.07.049. [DOI] [PubMed] [Google Scholar]
- Kaltoft K, Bisballe S, Dyrberg T, Boel E, Rasmussen PB, Thestrup-Pedersen K. Establishment of two continuous T-cell strains from a single plaque of a patient with mycosis fungoides. In Vitro Cell Dev Biol. 1992;28(3):161–7. doi: 10.1007/BF02631086. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Sahasrabuddhe AA, Rolland DCM, Velusamy T, Chung F, Schaller M, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sézary syndrome. Nature Communications. 2015 Sep 21;6:1–10. doi: 10.1038/ncomms9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Hess S, Richardson SK, Newton S. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798–812. doi: 10.1172/JCI24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch IR, Watanabe R, O’Malley JT, Williamson DW, Scott L-L, Elco CP, et al. TCR sequencing facilitates diagnosis and identifies mature T cells as the cell of origin in CTCL. Sci Transl Med. 2015 Oct 7;7(308):308ra158–8. doi: 10.1126/scitranslmed.aaa9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittipongdaja W, Wu X, Garner J, Liu X, Komas SM, Hwang ST, et al. Rapamycin Suppresses Tumor Growth and Alters the Metabolic Phenotype in T-Cell Lymphoma. J Invest Dermatol. 2015 May;7:1–8. doi: 10.1038/jid.2015.153. [DOI] [PubMed] [Google Scholar]
- Krejsgaard T, Kopp K, Ralfkiaer E, Willumsgaard AE, Eriksen KW, Labuda T, et al. A novel xenograft model of cutaneous T-cell lymphoma. Experimental Dermatology. 2010 Nov 19;19(12):1096–102. doi: 10.1111/j.1600-0625.2010.01138.x. [DOI] [PubMed] [Google Scholar]
- Krejsgaard T, Lindahl LM, Mongan NP, Wasik MA, Litvinov IV, Iversen L, et al. Malignant inflammation in cutaneous T cell lymphoma—a hostile takeover. Seminars in Immunopathology. 2017 Mar;20:1–14. doi: 10.1007/s00281-016-0594-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejsgaard T, Litvinov IV, Wang Y, Xia L, Willerslev-Olsen A, Koralov SB, et al. Elucidating the role of interleukin-17F in cutaneous T-cell lymphoma. Blood. 2013 Aug 8;122(6):943–50. doi: 10.1182/blood-2013-01-480889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejsgaard T, Willerslev-Olsen A, Lindahl LM, Bonefeld CM, Koralov SB, Geisler C, et al. Staphylococcal enterotoxins stimulate lymphoma-associated immune dysregulation. Blood. 2014 Jul 31;124(5):761–70. doi: 10.1182/blood-2014-01-551184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejsgaard TOR, Ralfkiaer U, Clasen-Linde E, Eriksen KW, Kopp KL, Bonefeld CM, et al. Malignant Cutaneous T-Cell Lymphoma Cells Express IL-17 Utilizing the Jak3-Stat3 Signaling Pathway. J Invest Dermatol. 2011 Feb 24;131(6):1331–8. doi: 10.1038/jid.2011.27. [DOI] [PubMed] [Google Scholar]
- Lamprecht B, Kreher S, Möbs M, Sterry W, Dörken B, Janz M, et al. The tumour suppressor p53 is frequently nonfunctional in Sézary syndrome. Br J Dermatol. 2012 Jun 6;167(2):240–6. doi: 10.1111/j.1365-2133.2012.10918.x. [DOI] [PubMed] [Google Scholar]
- Linnemann T, Gellrich S, Lukowsky A, Mielke A, Audring H, Sterry W, et al. Polyclonal expansion of T cells with the TCR Vbeta type of the tumour cell in lesions of cutaneous T-cell lymphoma: evidence for possible superantigen involvement. Br J Dermatol. 2004 May;150(5):1013–7. doi: 10.1111/j.1365-2133.2004.05970.x. [DOI] [PubMed] [Google Scholar]
- Litvinov IV, Tetzlaff MT, Rahme E, Habel Y, Risser DR, Gangar P, et al. Identification of geographic clustering and regions spared by cutaneous T-cell lymphoma in Texas using 2 distinct cancer registries. Cancer. 2015 Feb 27;121(12):1993–2003. doi: 10.1002/cncr.29301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood. 2015 Jul 23;126(4):508–19. doi: 10.1182/blood-2014-11-611194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie RCT, Jones CL, Tosi I, Caesar JA, Whittaker SJ, Mitchell TJ. Constitutive activation of STAT3 in Sézary syndrome is independent of SHP-1. Leukemia. 2011 Aug 5;26(2):323–31. doi: 10.1038/leu.2011.198. [DOI] [PubMed] [Google Scholar]
- Morales-Suárez-Varela MM, Olsen J, Johansen P, Kaerlev L, Guénel P, Arveux P, et al. Occupational Exposures and Mycosis Fungoides. A European Multicentre Case–control Study (Europe) Cancer Causes and Control. 2005 Dec;16(10):1253–9. doi: 10.1007/s10552-005-0456-6. [DOI] [PubMed] [Google Scholar]
- Moreau JF, Buchanich JM, Geskin JZ, Akilov OE, Geskin LJ. Non-random geographic distribution of patients with cutaneous T-cell lymphoma in the Greater Pittsburgh Area. Dermatol Online J. 2014 Jul;15:20. (7). [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, et al. Compartmentalized Control of Skin Immunity by Resident Commensals. Science. 2012 Aug 30;337(6098):1115–9. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netchiporouk E, Litvinov IV, Moreau L, Gilbert M, Sasseville D, Duvic M. Deregulation in STAT signaling is important for cutaneous T-cell lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle. 2014 Nov 14;13(21):3331–5. doi: 10.4161/15384101.2014.965061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V, Huggins RH, Lertsburapa T, Bauer K, Rademaker A, Gerami P, et al. Cutaneous T-cell lymphoma and Staphylococcus aureus colonization. J Am Acad Dermatol. 2008 Dec;59(6):949–52. doi: 10.1016/j.jaad.2008.08.030. [DOI] [PubMed] [Google Scholar]
- Pigozzi B, Bordignon M, Belloni Fortina A, Michelotto G, Alaibac M. Expression of the CD1a molecule in B- and T-lymphoproliferative skin conditions. Oncol Rep. 2006 Feb;15(2):347–51. [PubMed] [Google Scholar]
- Polk DB, Peek RM. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010 Jun;1:1–12. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro PE, Pinto FJ. The histologic spectrum of mycosis fungoides/Sézary syndrome (cutaneous T-cell lymphoma). A review of 222 biopsies, including newly described patterns and the earliest pathologic changes. Am J Surg Pathol. 1994 Jul;18(7):645–67. doi: 10.1097/00000478-199407000-00001. [DOI] [PubMed] [Google Scholar]
- Sommer VH, Clemmensen OJ, Nielsen O, Wasik M, Lovato P, Brender C, et al. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia. 2004 May 13;18(7):1288–95. doi: 10.1038/sj.leu.2403385. [DOI] [PubMed] [Google Scholar]
- Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci USA. 2005 Mar 29;102(13):4700–5. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005 Oct 25;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talpur R, Bassett R, Duvic M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sézary syndrome. Br J Dermatol. 2008 Jul;159(1):105–12. doi: 10.1111/j.1365-2133.2008.08612.x. [DOI] [PubMed] [Google Scholar]
- Thaler S, Burger AM, Schulz T, Brill B, Bittner A, Oberholzer PA, et al. Establishment of a mouse xenograft model for mycosis fungoides. Experimental Dermatology. 2004 Jul;13(7):406–12. doi: 10.1111/j.0906-6705.2004.00201.x. [DOI] [PubMed] [Google Scholar]
- Tokura Y, Yagi H, Ohshima A, Kurokawa S, Wakita H, Yokote R, et al. Cutaneous colonization with staphylococci influences the disease activity of Sézary syndrome: a potential role for bacterial superantigens. Br J Dermatol. 1995 Jul;133(1):6–12. doi: 10.1111/j.1365-2133.1995.tb02485.x. [DOI] [PubMed] [Google Scholar]
- van Doorn R, van Kester MS, Dijkman R, Vermeer MH. Oncogenomic analysis of mycosis fungoides reveals major differences with Sézary syndrome. Blood. 2009 doi: 10.1182/blood-2008-04-153031. [DOI] [PubMed] [Google Scholar]
- Verma NK, Davies AM, Long A, Kelleher D, Volkov Y. STAT3 knockdown by siRNA induces apoptosis in human cutaneous T-cell lymphoma line Hut78 via downregulation of Bcl-xL. Cell Mol Biol Lett. 2010 Jun;15(2):342–55. doi: 10.2478/s11658-010-0008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeer MH, van Doorn R, Dijkman R, Mao X, Whittaker S, van Voorst Vader PC, et al. Novel and Highly Recurrent Chromosomal Alterations in Sezary Syndrome. Cancer Research. 2008 Apr 15;68(8):2689–98. doi: 10.1158/0008-5472.CAN-07-6398. [DOI] [PubMed] [Google Scholar]
- Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, et al. Genomic profiling of Sézary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genetics. 2015 Nov 9;47(12):1426–34. doi: 10.1038/ng.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker SJ, Smith NP, Jones RR, Luzzatto L. Analysis of beta, gamma, and delta T-cell receptor genes in mycosis fungoides and Sézary syndrome. Cancer. 1991 Oct 1;68(7):1572–82. doi: 10.1002/1097-0142(19911001)68:7<1572::aid-cncr2820680719>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Willemze R. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005 May 15;105(10):3768–85. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- Willerslev-Olsen A, Krejsgaard T, Lindahl L, Bonefeld C, Wasik M, Koralov S, et al. Bacterial Toxins Fuel Disease Progression in Cutaneous T-Cell Lymphoma. Toxins. 2013 Aug;5(8):1402–21. doi: 10.3390/toxins5081402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HK, Mishra A, Hake T, Porcu P. Evolving Insights in the Pathogenesis and Therapy of Cutaneous T-cell lymphoma (Mycosis Fungoides and Sézary Syndrome) Br J Haematol. 2011 Aug 25;155(2):150–66. doi: 10.1111/j.1365-2141.2011.08852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woollard WJ, Pullabhatla V, Lorenc A, Patel VM, Butler RM, Bayega A, et al. Candidate driver genes involved in genome maintenance and DNA repair in Sézary syndrome. Blood. 2016 Jun 30;127(26):3387–97. doi: 10.1182/blood-2016-02-699843. [DOI] [PubMed] [Google Scholar]
- Wu X, Sells RE, Hwang ST. Upregulation of Inflammatory Cytokines and Oncogenic Signal Pathways Preceding Tumor Formation in a Murine Model of T-Cell Lymphoma in Skin. J Invest Dermatol. 2011 Apr 14;131(8):1727–34. doi: 10.1038/jid.2011.89. [DOI] [PubMed] [Google Scholar]
- Wu X, Wang TW, Lessmann GM, Saleh J, Liu X, Chitambar CR, et al. Gallium Maltolate Inhibits Human Cutaneous T-Cell Lymphoma Tumor Development in Mice. J Invest Dermatol. 2014 Dec 11;135(3):877–84. doi: 10.1038/jid.2014.476. [DOI] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T Helper 17 Lineage Differentiation Is Programmed by Orphan Nuclear Receptors RORα and RORγ. Immunity. 2008 Jan;28(1):29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.