




Abstract
Neurofibromatosis type 1 (NF1) is a common genetic disorder that predisposes affected individuals to tumours. The NF1 gene encodes a RAS GTPase-activating protein called neurofibromin and is one of several genes that (when mutant) affect RAS–MAPK signalling, causing related diseases collectively known as RASopathies. Several RASopathies, beyond NF1, are cancer predisposition syndromes. Somatic NF1 mutations also occur in 5–10% of human sporadic cancers and may contribute to resistance to therapy. To highlight areas for investigation in RASopathies and sporadic tumours with NF1 mutations, we summarize current knowledge of NF1 disease, the NF1 gene and neurofibromin, neurofibromin signalling pathways and recent developments in NF1 therapeutics.
Neurofibromatosis type 1 (NF1; Online Mendelian Inheritance in Man (OMIM) database 162200) is a common inherited tumour predisposition syndrome that affects approximately 1 in 3,000 individuals worldwide1–3. The history of NF1 research and NF1 diagnostic criteria are described in FIG. 1. Some manifestations of NF1 are observed in early childhood, whereas others present later in life (FIG. 2).
Figure 1. Neurofibromatosis type 1 historical developments.
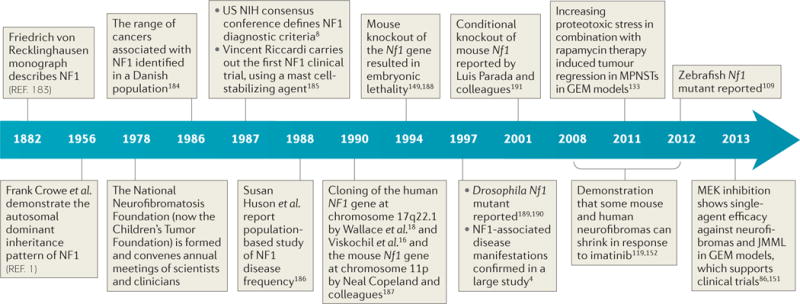
From the development of diagnostic criteria to the development of ongoing clinical trials, the neurofibromatosis type 1 (NF1) field has been aided by close clinician–scientist interactions, which have been facilitated by the Children’s Tumor Foundation. Currently accepted diagnostic criteria include six or more café-au-lait macules with a minimum diameter of >5 mm in pre-pubertal subjects; two or more neurofibromas of any type or one plexiform neurofibroma; freckling in the axillary or inguinal region; optic pathway glioma; two or more Lisch nodules (iris hamartomas); a distinctive osseous lesion, such as sphenoid dysplasia or thinning of long bone cortex with or without pseudarthrosis; and a first-degree relative with NF1 according to these criteria. GEM, genetically engineered mouse; JMML, juvenile myelomonocytic leukaemia; MPNSTs, malignant peripheral nerve sheath tumours; NIH, National Institutes of Health.
Figure 2. Disease manifestations in patients with neurofibromatosis type 1: epochs in which they develop.
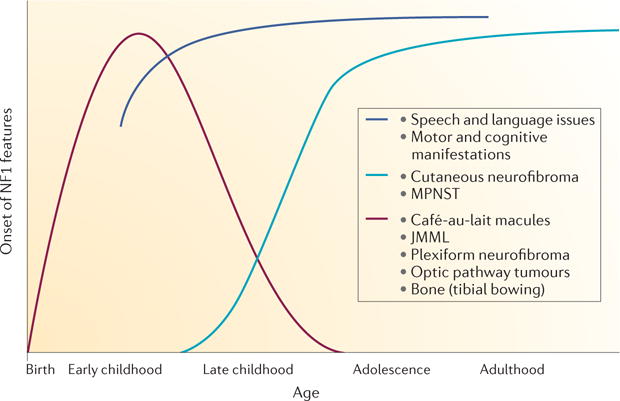
Most plexiform neurofibromas are present at a very young age but, depending on the tumour location, may not be diagnosed until later in life if whole-body magnetic resonance imaging is not performed192. Multiple hyperpigmented skin lesions (café-au-lait macules) are an early sign of neurofibromatosis type 1 (NF1) and are observable in children under 3 years of age193. Young children may also present with bone dysplasia194, delayed speech195 and delayed acquisition of motor skills196. Young children with NF1 are at an increased risk of developing juvenile myelomonocytic leukaemia (JMML)10 and optic pathway glioma (mean age of 5 years197). Later in childhood, cognitive issues surface198. If the features labelled in burgundy do not occur early, they will not develop later in life. The dark blue line shows that speech, language, motor and cognitive changes are detected, as children would normally develop specific skills. Cutaneous neurofibromas typically begin to grow during puberty5. Although malignant peripheral nerve sheath tumours (MPNSTs) may occur in childhood, they are most common in adult patients with NF1 over 30 years of age124. Beyond cancer, it is now appreciated that generalized or specific cognitive impairment is observed in >50% of patients with NF1 (REF. 198), and many have attention deficit hyperactivity disorder. Features of autism spectrum disorder may occur199, and vascular defects are common200. On the basis of cloning of the NF1 gene, understanding of the related disorders and clarification of age-of-onset of individual disease manifestations, it has been suggested (since 2007) that the diagnostic criteria may need revision201.
Almost all patients with NF1 develop cutaneous neurofibromas, which are benign peripheral nerve tumours4,5. Some patients also develop benign plexiform neurofibromas, which can cause substantial morbidity and can degenerate to form peripheral nerve sarcomas known as malignant peripheral nerve sheath tumours (MPNSTs). These tumours are key contributors to reduced life expectancy in NF1 (REF. 6). Another common tumour in patients with NF1 is optic pathway glioma (OPG)7. In 1988, a US National Institutes of Health (NIH) consensus conference defined the currently used NF1 diagnostic criteria8,9. Notably, these criteria include neurofibroma and OPG but do not include malignant disease. Rarer tumours that develop in patients with NF1 are juvenile myelomonocytic leukaemia (JMML)10, benign or malignant pheochromocytoma11, gastrointestinal stromal tumour (GIST)12, glomus tumours13, juvenile xanthogranuloma, rhabdomyosarcoma14 and lipoma15. Cloning of the NF1 gene (OMIM 613113) led to the identification of biallelic NF1 mutations in patient-derived tumours, which in turn immediately led to classification of NF1 as a tumour suppressor gene16–18. All NF1-related tumours show biallelic inactivation of the NF1 gene19,20. Patients with NF1 may also be at an increased risk of developing secondary cancers following radiation exposure, and it is important to consider this risk in the treatment of this predisposed population21. In addition, patients with NF1 have an increased risk of developing several adult cancers22. An analysis of UK death certificates found that patients with NF1 may also be at an increased risk of cancers of the gastrointestinal tract, liver, lung, bone, thyroid, breast and ovary22. Risk of malignant melanoma, non-Hodgkin lymphoma and chronic myeloid leukaemia might also be increased.
Early studies identified the protein encoded by NF1, neurofibromin, as having homology to the yeast proteins Ira1 and Ira2, which are inhibitory regulators of the RAS–cyclic AMP pathway23–25. In yeast, Ira proteins negatively regulate Ras by converting it from the active GTP-bound form to the inactive GDP-bound form. This is required to reduce levels of cAMP under nutrient-limiting conditions and to mediate membrane association of adenylyl cyclase. Neurofibromin is a GTPase-activating protein (GAP) that regulates RAS (RASGAP). It binds to GTP-bound RAS through its GAP-related domain (GRD) to dramatically augment its intrinsic GTPase activity26. Neurofibromin thereby functions as an off signal for all of the vertebrate RAS GTPases, including HRAS, NRAS, KRAS, MRAS, RRAS and RRAS2 (also known as TC21)27. Therefore, loss of NF1 activates signalling through the RAS pathway, which is a key driver of cancer. GTP-bound RAS activates multiple effector pathways, including the RAS–MAPK pathway, in which GTP-bound RAS activates the RAF–MEK–ERK cascade (reviewed in REF. 28) (FIG. 3).
Figure 3. Neurofibromatosis type 1 signalling pathways.
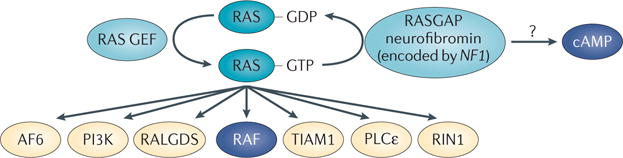
In the absence of negative regulation of RAS proteins, resulting from loss of neurofibromatosis type 1 (NF1, which encodes neurofibromin), GTP-bound RAS levels are increased. Therefore, signalling pathways downstream of RAS that are normally activated by receptors — including receptor tyrosine kinases, integrins and ion channels — show enhanced activation. RAS signalling pathways include the MEK–ERK signalling cascade downstream of RAF and also many other potential RAS effectors, including AF6, an F-actin and RAP1-binding protein; RAL guanine nucleotide dissociation stimulator (RALGDS), a guanine nucleotide exchange factor (GEF) for the RALA and RALB GTPases; T lymphoma invasion and metastasis-inducing protein 1 (TIAM1), an exchange factor for the GTPase RAC1; phospholipase Cε (PLCε), an isoform of the phospholipase C family; and RAS and RAB interactor 1 (RIN1), which is a RAS effector and RAB5 GEF. In addition, loss of NF1 results in deregulation of cyclic AMP levels in affected cells through poorly characterized mechanisms that may be independent of RAS and/or result from crosstalk between RAS and heterotrimeric G protein signalling. RAF and cAMP are the only effector pathways currently shown to have therapeutic potential in NF1 disease. GAP, GTPase-activating protein.
Mutations in many other genes that encode components of the RAS–MAPK pathway also predispose patients to partially overlapping sets of manifestations (known as RASopathies), which can include tumours29 (BOX 1). Patients with Noonan syndrome have a 4% risk of developing cancer by the age of 20 years, with JMML predominating and rhabdomyosarcoma, neuroblastoma and low-grade glioma occurring at lower incidences30. Patients with Costello syndrome have a 15% risk of cancer by the age of 20 years, with rhabdomyosarcoma developing in 9% of patients, and neuroblastoma and bladder cancer developing in 1% of patients30. Patients with Legius syndrome develop lipoma31. The fact that all known RASopathy mutations affect the RAS–MAPK signalling pathway supports the idea that the MAPK pathway downstream of RAS, and not other pathways downstream of RAS, is the crucial driver of tumorigenesis in patients with RASopathies. ‘RASopathy clinics’ are beginning to study and compare patients with RASopathies. It is hoped that some or all RASopathy manifestations will respond to therapies such as MAPK inhibitors.
Importantly, sequencing of tumour exomes and genomes has revealed that somatic NF1 mutations are present at incidences from 2.5% to 11.8% in sporadic, predominantly adult, tumour types such as lung cancer32, glioblastoma33, ovarian cancer34, breast cancer35 and acute myeloid leukaemia (AML)36; however, they are also present in the paediatric tumour rhabdomyosarcoma.
NF1 genetics: mutations and modifiers
Structure of the NF1 gene and mutational analysis
The NF1 gene contains 60 exons and generates multiple alternatively spliced isoforms37. More than 1,400 mutations in the NF1 gene have been reported in the Human Gene Mutation Database, most of which are clearly loss-of-function alleles. These include splice site, nonsense and missense mutations, as well as deletions, insertions, frameshifts and translocations38. Notably, several patient missense mutations that affect the neurofibromin GRD selectively diminish GAP activity, which supports the notion that the regulation of RAS has a crucial role in NF1 disease39. Identification of NF1 mutations in patients remains difficult owing to the large gene size and structure, as well as the large range of mutations that have been identified40. For many years, 95% of patient mutations were identified using a combination of complementary methods, including protein truncation, fluorescence in situ hybridization, heteroduplex, Southern blot and cytogenetic analyses38. A preliminary report applied next-generation sequencing to samples from patients with NF1 (REF. 41), and DNA-based sequencing is now being offered as a clinical test for diagnostic purposes (see the website of University of Alabama at Birmingham Medical Genomics Laboratory). Thus, NF1 mutation analysis can assist in diagnosing cases of NF1 in which a clinical diagnosis cannot be established with certainty.
The variability in manifestations in patients from a single family with the same NF1 mutation does not support a major role for genotype–phenotype correlations in NF1 (REF. 42), although there are several important exceptions. Germline splice site mutations occur in 30% of patients with NF1, and these patients may have an increased overall tumour risk43. Mutations that delete the NF1 gene, and several flanking genes, occur in up to 10% of patients with NF1. This class of mutations predisposes affected individuals to an increased risk of intellectual disability, to greater numbers of cutaneous neurofibromas and to MPNSTs44. Another genotype–phenotype correlation is of a very rare 3-bp deletion in patients with NF1 who lack neurofibromas45.
Modifier genes in NF1
Given the paucity of NF1 genotype–phenotype correlations, it was proposed that modifier genes underlie the variable penetrance of NF1. Monozygotic twins with NF1 showed a high degree of concordance for cutaneous neurofibroma tumour burden and numbers of café-au-lait macules, supporting the idea that modifier genes contribute to these features46,47. The large polygenic deletions (mentioned above) indicate that modifier genes might be linked to NF1 (REF. 44). Recently, the gene encoding the chromatin remodelling complex Polycomb repressive complex 2 subunit SUZ12, which lies within this region, has been shown to be a cooperating tumour suppressor in mouse models and in human tumours48–50. Studies in mouse models also support roles for modifier genes in NF1. Astrocytoma resistance alleles were recently identified as spinal cord resistance to astrocytoma modifier 1 (Scram1) and astrocytoma resistance locus in males 1 (Arlm1) loci38,51,52. It is unclear whether disease modifier genes in NF1 are also relevant in NF1-mutant sporadic tumours.
In support of a role for modifier genes in NF1 is OPGs; these tumours have a decreased prevalence in the African-American population compared with other races53. Sex-linked factors may modify prognosis; males are at an increased risk of sporadic high-grade glioma, but NF1 females with low-grade OPG have a worse prognosis than males with NF1-related OPG54. In addition, male Nf1−/− mouse astrocytes expressing dominant-negative p53 show increased tumorigenesis and inactivation of the RB protein compared with cells derived from females55. An imprinting control region remotely interacts with an intergenic sequence between Nf1 and Wsb1 on chromosome 11 to regulate Nf1 transcription, and mutations in this intergenic sequence could also potentially modify NF1 disease56.
NF1 mutations in sporadic cancers
The advancement of whole-genome sequencing has resulted in the identification of NF1 mutations in various non-NF1-associated sporadic cancers, including glioblastoma33,57, neuroblastoma58, AML36, lung cancer32, ovarian cancer34 and breast cancer59. We anticipate that the identification of tumours that contain NF1 mutations will continue to increase with future sequencing efforts. A comprehensive study that analysed somatic mutation patterns in more than 1,500 cancer-related genes in a large panel of lung, breast, ovarian, pancreatic and prostate tumours identified NF1 (mutation frequency >5%) as one of ten genes that are mutated most often in these types of tumours. In comparison, the point mutation frequency was 33% for TP53, 7% for KRAS and 5% for cyclin-dependent kinase inhibitor 2A (CDKN2A)59; however, deletions in CDKN2A are much more common than deletions in NF1. It is as yet unknown whether biallelic loss of NF1 is common or whether only hemizygous loss of NF1 contributes to tumour progression in sporadic disease. Consistent with the latter possibility, mouse cells hemizygous for Nf1 mutations show abnormal growth and invasion60–62. Hemizygous NF1-mutant cells might show lower levels of GTP-bound RAS than cells with complete inactivation of NF1 and/or develop mutations in additional RAS–MAPK pathway genes to affect tumour properties. Although sporadic tumours with NF1 mutations are largely exclusive of those tumours that harbour mutations in MAPK kinase 1 (MAP2K1) or NRAS, on the basis of our analyses of somatic co-mutation patterns in The Cancer Genome Atlas data sets (cBio Portal for Cancer Genomics), 11 of 114 melanomas with NF1 mutations also show mutations in BRAF, NRAS or RAF1. Thus, there may be subcategories of tumours, and perhaps cells, in which BRAF, NRAS or RAF1 are co-mutated with NF1.
In tumours with NF1 mutations, the order of mutations seems to affect tumour grade in specific cell types; for example, initial loss of NF1 in nerve glial cells triggers neurofibroma. In this case, oncogene-induced senescence occurs, and inactivation of p53 bypasses this response for progression to MPNSTs63. Similarly, benign grade 1 astrocytomas develop in genetically engineered mice (GEMs) when Nf1 is lost first64. By contrast, aggressive gliomas form when Trp53 is co-mutated with Nf1 (REF. 65). Indeed, in human glioblastomas, half of the tumours with NF1 mutations also harbour TP53 mutations66. Mutational order may explain why patients with NF1 are not predisposed to certain sporadic tumours, such as lung tumours, whereas >10% of sporadic lung cancers32 have NF1 mutations, which are probably acquired late in tumorigenesis.
The NF1 protein: neurofibromin
Neurofibromin is a large multi-domain 2,818 amino acid protein67. Exon 23 encodes part of the GRD, which is the RAS regulatory domain of neurofibromin. An exon 23 splice variant inserts an alternative exon 23a, which decreases neurofibromin RASGAP activity68. In addition to this central GRD, neurofibromin contains other functional domains, most of which are of uncertain importance in the tumour suppressor function of neurofibromin (FIG. 4).
Figure 4. Neurofibromin protein structure and interacting proteins.
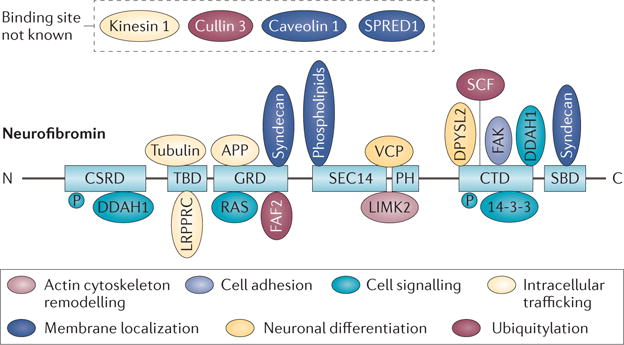
Neurofibromin contains multiple domains (light blue). These include a cysteine–serine-rich domain (CSRD), a tubulin-binding domain (TBD), a central GTPase-activating protein-related domain (GRD), a SEC14 domain202,203, a pleckstrin homology (PH) domain, a carboxy-terminal domain (CTD) and a syndecan-binding domain (SBD). The SEC14 and PH domains bind to phospholipids, and have been studied structurally204. Proteins identified as neurofibromin-interacting proteins and phospholipids (ovals) are shown associated with functions ascribed to them, including intracellular trafficking (light yellow); neuronal differentiation (dark yellow); membrane localization (dark blue); actin cytoskeleton remodelling (light pink); ubiquitylation (dark pink); cell adhesion (purple) and cell signalling through nitric oxide via dimethylarginine dimethylaminohydrolase 1 (DDAH1) and RAS (turquoise). Each interacting protein is shown bound to the domain of neurofibromin with which it is believed to interact. Some proteins are known to interact with neurofibromin, but the binding site is unknown. Phosphorylation (P) sites implicated as protein kinase A substrates are shown. Descriptions of each interacting protein, binding domains and literature references are shown in Supplementary information S1 (table). APP, amyloid-β (A4) precursor protein; DPYSL2, dihydropyrimidinase-related protein 2; FAF2, FAS-associated factor 2; FAK, focal adhesion kinase; LIMK2, LIM domain kinase 2; LRPPRC, leucine-rich pentatricopeptide motif-containing protein; SCF, Skp, Cullin, F-box-containing complex; SPRED1, sprouty-related, EVH1 domain-containing protein 1; VCP, valosin-containing protein.
Interactions of neurofibromin
In 1991, Bollag and McCormick69 reported that the lipids arachidonate, phosphatidate and phosphatidylinositol-4,5-bisphosphate inhibit neurofibromin GAP activity, but no definitive in vivo role for lipid–neurofibromin interaction through the SEC14 and pleckstrin homology (PH) domains has so far been identified. Neurofibromin is also implicated in connecting RAS signalling to activation of RHO-family GTPases, which results in modulation of the cytoskeleton. Therefore, it is intriguing that the neurofibromin phospholipid-binding SEC14 and PH domains interact with LIM domain kinase 2 (LIMK2) and thereby inhibit activation of LIMK2 by RHO-associated protein kinase, which is known to modulate the actin cytoskeleton70. Some neurofibromin domains may function as scaffolds that target neurofibromin to specific intracellular locations. Indeed, neurofibromin interacts with sprouty-related, EVH1 domain-containing protein 1 (SPRED1). This interaction results in localization of neurofibromin to membranes, which enables neurofibromin to down-regulate GTP-bound RAS71. Interestingly, SPRED1 is also a RASopathy gene. Unlike mutations in NF1, mutations in SPRED1 do not predispose affected individuals to neurofibromas, gliomas or MPNSTs but rather to lipomas. Distinct features of each disorder may arise from cell type-specific use of components of the MAPK pathway.
Neurofibromin is the main RASGAP in neuronal dendritic spines72, where the molecular chaperone valosin-containing protein (VCP) interacts with the leucine-rich repeat domain of neurofibromin. In mice, mutations in Vcp, like loss of Nf1, reduce spine density73. Importantly, VCP functions downstream of neurofibromin, and expression of VCP rescues spine defects in Nf1+/− neurons. VCP is now under investigation as a cancer target. Dimethylarginine dimethylaminohydrolase 1 (DDAH1), which degrades the endogenous nitric oxide inhibitor asymmetric dimethylarginine (ADMA), was identified as another neurofibromin interaction partner. Knockdown of Nf1 rescued the decrease in cell proliferation caused by knockdown of Ddah1 in mouse endothelial cells74,75, and this is of special interest because the modulation of nitric oxide is an attractive therapeutic target.
Neurofibromin regulation and signalling
Neurofibromin protein levels can also be affected by mechanisms beyond NF1 mutation. One category of neurofibromin-interacting proteins is ubiquitin ligases, which ubiquitylate and cause degradation of neurofibromin, thus sustaining RAS signalling. The ubiquitin ligase SAG (sensitive to apoptosis gene protein; also known as RNF7 and RBX2) was reported to interact with neurofibromin and affect vascular development76, whereas the ubiquitin ligase cullin 3 degraded neurofibromin in glioblastoma cells in which neurofibromin was not mutated76,77. It remains to be determined whether these and/or other ubiquitin ligases only degrade neurofibromin in specific settings. Downregulation of neurofibromin may also result from the methylation of NF1 in cancer cells78,79. NF1 is also a target of micro-RNAs (mi RNAs); expression of miR-128 in neurons and miR-193b in head and neck squamous cell carcinoma cells decreased the levels of NF1 mRNA and neurofibromin80,81.
Modulation of RAS signalling by neurofibromin
The absence of neurofibromin leads to slowed hydrolysis of GTP-bound RAS, which sustains RAS signalling. Nevertheless, upstream receptors are required to activate RAS, and several receptors have been implicated upstream of neurofibromin in particular cell types. For example, genetic and biochemical evidence support a necessary role of granulocyte–macrophage colony-stimulating factor (GM-CSF) and activation of the GM-CSF receptor to maintain JMML in Nf1-mutant mice82. This receptor may also have a role in neurofibroma formation after nerve injury83. By contrast, activation of the KIT receptor by stem cell factor (also known as KIT ligand) has a key role in neurofibroma formation in mice62. In Drosophila melanogaster expressing mutant Nf1, the loss of the receptor tyrosine kinase Anaplastic lymphoma kinase (Alk) rescues the small size of the flies and ERK activation84. Other receptors probably function as upstream regulators of neurofibromin in specific cell types.
Neurofibromin and downstream signalling
Of the many putative downstream effectors of RAS signalling — RAL guanine nucleotide dissociation stimulator (RALGDS), PI3K, phospholipase Cε, T lymphoma invasion and metastasis-inducing protein 1 (TIAM1), RAS association domain family protein (RASSF), RAS and RAB interactor 1 (RIN1), RIN2, RIN3, AF6 (also known as afadin and MLLT4), impedes mitogenic signal propagation (IMP; also known as BRAP2), RAS-associated and PH domains-containing protein 1 (RAPH1), growth factor receptor-bound protein 7 (GRB7), and PDZGEF1 (also known as RAPGEF2) (FIG. 2) — only a few have been studied in the context of NF1. The best studied is the neurofibromin–RAS–MAPK pathway. Loss of neurofibromin results in the activation of this pathway in multiple types of tumour85,86. Heat shock response87 and MAF regulation of mTOR signalling88 also occur downstream of NF1 loss and subsequent activation of ERK. RAL guanine nucleotide exchange factors have also been implicated downstream of RAS in MPNSTs89.
Several studies in mice support important roles for PI3K signalling when loss of Nf1 drives tumour formation, and several pathways have been implicated downstream of PI3K. For example, PI3K–mTOR signalling is enhanced in Nf1-mutant astrocytes and MPNSTs90,91. PI3K–AKT signalling downstream of Rras2 has a role in the initiation of neurofibromas92; activation of the PI3K pathway through loss of Pten in mice promotes the transformation of Nf1-driven neurofibromas to MPNSTs93. Sustained activation of AKT in MPNST cells requires calcium and calmodulin94. On the basis of these results, it will be important to consider cell type-specific neurofibromin–RAS effector pathways when attempting to identify therapeutic targets. In addition, simultaneously targeting multiple RAS effector pathways may provide enhanced effects.
NF1 and cAMP
In yeast, the NF1 homologues IRA1 (REF. 95) and IRA2 (REF. 96) regulate both Ras and cAMP signalling. Ira1 and Ira2 each simultaneously bind to Ras2 and adenylyl cyclase, thus regulating both pathways97. It is accepted that increased levels of cAMP inhibit the proliferation of most cell types, and it has been proposed that altered levels of cAMP contribute to cancer. Mouse98, D. melanogaster 99 and zebrafish100 cells expressing mutant Nf1 all show deregulated cAMP levels. In D. melanogaster lacking Nf1 cAMP levels are low, but it remains unclear whether this results from increased Ras activity or whether it occurs independently of Ras98,99. By contrast, cAMP levels are increased in mouse Schwann cells (peripheral nerve support cells) or human MPNST cells lacking NF1 (REF. 101). Many different RAS–cAMP pathway interactions may occur in specific NF1-mutant cell types. In mouse neurons, atypical protein kinase C-mediated β-adrenergic receptor kinase 1 (ADRBK1)-driven inactivation of the G protein Gαs modulated RAS activity98,102 . In another system, the protein kinase A-activated transcription factor cAMP-responsive element-binding protein (CREB) bound to the mir-9 promoter, which repressed expression of NF1 and encouraged cell migration103. Interfering with cAMP signalling may present a therapeutic opportunity in several manifestations of NF1, as was proposed for NF1-driven brain tumours104. Blocking RAS–MAPK signalling and increasing cAMP levels may be useful therapeutically; for example, reversing one zebrafish brain defect required blockade of MEK, whereas reversing another defect required an increase in cAMP levels100.
NF1 tumorigenesis and tumour cells of origin
Many tumours in patients with germline NF1 mutations are neural crest cell-derived tumours (pheochromocytomas, neurofibromas and MPNSTs) or neuroepithelial cell-derived tumours (pilocytic astrocytomas). Non-neural-crest-related cells are also predisposed to tumorigenesis (JMML and rhabdomyosarcoma). It is not known why specific cell types are sensitive to loss of NF1. Affected populations do not express increased levels of neurofibromin compared with other cells; it has been speculated that these populations critically rely on neurofibromin rather than other GAPs.
Low-grade astrocytoma
At least 15% of patients with NF1 develop OPGs, which are mainly grade I pilocytic astrocytomas7. These tumours are defined as benign, generally have a favourable prognosis and rarely progress105. In contrast to NF1-related pilocytic astrocytomas, pilocytic astrocytomas in patients without NF1 are typically more aggressive, although they have RAS pathway mutations, including BRAF duplications or activating point mutations in BRAF or KRAS106. In GEM models of grade 1 astrocytoma resulting from loss of Nf1, many neurons in the brain and most macroglial cells are Nf1−/− owing to use of glial fibrillary acidic protein (Gfap)–Cre or brain lipid-binding protein (Blbp; also known as Fabp7)–Cre drivers, which excise Nf1 in most brain stem and progenitor cells64,98,107. In this model, other cells of the body are Nf1+/− (REF. 64). Evidence indicates that when progenitor cells of the developing third ventricle lack Nf1, they develop into aberrant astrocytes, which are characteristic of pilocytic astrocytoma108. Consistent with this interpretation, loss of Nf1 in other brain cell types (such as oligodendrocytes or NG2 cells) failed to generate astrocytomas in zebrafish or mice109,110.
Cutaneous neurofibroma
All neurofibromas contain nerve Schwann cells (with biallelic NF1 mutations111), as well as NF1 wild-type or heterozygous fibroblasts, mast cells, macrophages, perineurial cells and endothelial cells. The percentage of each cell type varies from tumour to tumour. Neurofibromas in the human dermis or epidermis (known as cutaneous or dermal neurofibromas) are benign and do not transform; however, they can cause a substantial cosmetic burden in patients. It has been a source of confusion that some plexiform neurofibromas, which are more aggressive than cutaneous neurofibromas, also develop in the skin and subcutaneous tissue; alternative nomenclature has been discussed but not defined by consensus. There are no model organisms at present in which cutaneous neurofibromas spontaneously develop. However, after growth in vitro and transplantation into Nf1+/− syngeneic hosts, skin hair follicle-derived Nf1−/− precursors (SKPs) formed tumours that resembled dermal neurofibromas112. Transplantation of Nf1−/− SKPs into pregnant female mice increased growth of neurofibromas in the mouse model112, and cutaneous neurofibromas can develop and grow during puberty5 and can increase in size and number during pregnancy113. However, it remains unclear whether and which hormones or other factors increase the growth of human cutaneous neurofibromas. It is also unclear which factors limit the growth of most dermal neurofibromas in humans, and why they are resistant to transformation.
Plexiform neurofibroma
Plexiform neurofibromas are complex tumours that can weigh kilograms and can compress vital structures. Therefore, they are of considerable interest as targets of therapy. Plexiform neurofibromas develop within peripheral nerves and their perineurial sheaths. However, plexiform neurofibromas can invade adjacent tissue by disrupting the perineurium, remaining non-metastatic and clinically ‘benign’ but accounting for substantial morbidity and an increased risk of mortality when symptomatic114.
Extensive modelling of plexiform neurofibromas has been carried out in mice. Plexiform neurofibroma models all have biallelic loss of Nf1 in the Schwann cell lineage115–118, which is driven by P0 (also known as myelin protein zero (Mpz))–Cre, desert hedgehog (Dhh)–Cre, tamoxifen-inducible proteolipid protein (myelin) 1 (Plp1)–Cre, or Krox20 (also known as Egr2)–Cre. Each of these drivers knock out expression in Schwann cell progenitors, with differences in precise timing, location and the number of cells affected. Some models, probably those with fewer cells showing recombination, require additional hemizygous inactivation of Nf1 in haematopoietic cells to generate neurofibromas119. In all cases, the tumours (grade 1 neurofibromas) in GEM models resemble those of humans. In humans, plexiform neurofibromas primarily develop very early in life. Unexpectedly, plexiform neurofibromas can be induced even in adult mice, although in one model these are rare116,117. Unfortunately, this result does not resolve the uncertainty around the plexiform neurofibroma cell of origin, as adult mouse dorsal root ganglia and dorsal roots contain stem-like cells, mature Schwann cells and satellite cells, all of which have been proposed as possible neurofibroma-initiating cells115–118,120. Epidermal growth factor receptor (EGFR)-expressing Schwann cell precursor-like cells are present in mouse and human neurofibromas, and form neurofibroma-like lesions under the skin120 or in peripheral nerves121 of immunocompromised mice. Extensively passaged Schwann cells from neurofibromas can also form neurofibromas in immunocompromised mice122. Thus, de-differentiated Schwann cells and/or Schwann cell precursors may each be cells of origin for neurofibromas.
MPNSTs
MPNSTs are nerve-associated sarcomas, most of which arise in pre-existing plexiform neurofibromas in patients with NF1 (REF. 123), and are aggressive tumours that typically metastasize to the brain, bone and other sites124. Half of all MPNSTs arise sporadically, and the other half occur in patients with NF1 (REF. 125). Genetic alterations that affect the RAS pathway, including NF1, BRAF, NRAS or KRAS mutations, have been identified in some sporadic MPNSTs126,127, and NF1 and sporadic MPNSTs share a gene signature128. MPNSTs are also typically hyperdiploid, and their genomes — like genomes in most sarcomas — are highly rearranged129. Mutations associated with the transformation from benign plexiform neurofibromas to malignant MPNSTs include early mutations in CDKN2A129 and later mutations in TP53 (REFS 130–132) and SUZ12 (REF. 133). Loss of the tumour suppressor gene RB1 (which encodes RB) is found in 25% of MPNSTs134,135, and monosomy for the PTEN locus is observed in 50% of MPNSTs126,134,136,137. Low-level amplification of growth factor receptor genes, including EGFR, is also common126.
Human MPNSTs and MPNST cell lines contain CD133+ cells that may be stem-like cells138,139. GEM models of MPNSTs include combined loss of Nf1 and Trp53 (REFS 140,141) or Nf1 and Cdkn2a142,143. MPNSTs from these models and human MPNST cell lines contain stem-like cells that propagate disease144. Comparative oncogenomics and insertional mutagenesis screens have been used to identify candidate drivers of MPNST formation, including MEK, β-catenin and embryonic stem cell-expressed RAS (ERAS)86,145,146. In zebrafish, ribosomal gene mutations predispose to the formation of MPNSTs, and resulting tumours show loss of p53 translation147.
Other tumours in patients with NF1
Pheochromocytomas11, rhabdomyosarcomas14, glomus tumours20,148 and GISTs12 are present at increased incidence in patients with NF1 and show biallelic inactivation of NF1. Of these, only pheochromocytoma has been successfully modelled to date. Pheochromocytomas form in 15% of Nf1+/− mice149; however, this model has not been used in preclinical tests, probably owing to low tumour incidence. JMML is a rare paediatric manifestation of NF1 (REF. 10) and a RAS-driven haematopoietic stem cell disorder. Thus, patients with NF1 and patients with RASopathies who have mutations in NRAS, KRAS, CBL, protein tyrosine phosphatase non-receptor type 11 (PTPN11; also known as SHP2) or son of sevenless homologue 1 (SOS1) are predisposed to this low-grade leukaemia150, which is recapitulated in an Mx1–Cre;Nf1fl/fl model151.
Therapeutic implications
Preclinical testing
Importantly, the available mouse models of JMML, OPG, plexiform neurofibroma and MPNST are currently used for preclinical testing. Consistent with a major role for RAS–MAPK signalling in JMML, preclinical testing showed significant response to inhibition of MEK151. Disease burden was markedly reduced, although mutant stem cells persisted. The finding that the multi-receptor tyrosine kinase inhibitor imatinib or inhibition of MEK can shrink plexiform neurofibromas in mouse models led to clinical trials86,119,151. In the imatinib trial, 6 of 36 patients, primarily young children with small plexiform neurofibromas, responded to treatment; the target kinase (or kinases) affected in these patients is not yet known152. In a Phase I trial of the MEK inhibitor selumetinib (presented at the 2014 American Society of Clinical Oncology meeting), 11 of 18 patients with plexiform neurofibromas, some up to kilograms in weight, shrank by ≥20% in response to therapy, and many showed prolonged response153. These positive results not only support continued use of mouse models to guide clinical testing in human neurofibromas but also emphasize the finding that these benign tumours can respond to single-agent therapy for up to 3 years without showing resistance.
Complete surgical resection is required to cure MPNSTs, and no single-agent or combination tested to date has cured MPNSTs in any model system (reviewed in REF. 154). Consistent with the idea that combination therapy will be necessary to treat these aggressive cancers, the mTOR complex 1 (mTORC1) inhibitor rapamycin, together with agents that enhance oxidative stress, shrank MPNSTs133. In MPNST xenografts, prolonged responses have been observed with Aurora kinase inhibition and bromodomain inhibitors, but these have not yet been tested in mouse models of MPNSTs or in patients with NF1 (REFS 155,156). In some MPNSTs, autocrine chemokine (C-X-C motif) ligand 12 (CXCL12)–CXC receptor 4 (CXCR4) signalling activates β-catenin through the AKT–glycogen synthase kinase 3β–β-catenin stabilization pathway157. Transposon mutagenesis confirmed WNT pathway signalling as a driver of the transformation to MPNSTs, and many β-catenin pathway genes are deregulated in neurofibroma and MPNSTs158. Although other links between NF1 signalling and β-catenin pathway activation remain to be identified, these data and analysis of human NF1 Schwann cells support inhibition of the β-catenin pathway as a possible therapeutic target in NF1-driven tumours159.
NF1 loss as a drug resistance marker
MPNSTs in patients with NF1 are notoriously resistant to chemotherapy and radiation therapy. Recently, a large-scale study showed almost no benefit of chemotherapy or radiotherapy for NF1 MPNSTs, and a worse outcome has been reported for NF1 than for sporadic MPNSTs124. These data must be interpreted with caution, as late diagnosis of MPNSTs arising in plexiform neurofibromas in patients with NF1 may account for differences between the groups in treatment outcome160. However, although reasons might differ as to what causes resistance in sporadic tumours, NF1 has been identified as a gene that confers resistance to targeted therapy, including inhibition of kinases and the RAS pathway, in sporadic neuroblastoma58, lung carcinoma161 and melanoma162. In lung cancer models, resistance to EGFR therapy was mediated by NF1, and blocking MEK restored the response161. Melanoma develops in mice with mutant Braf, and loss of Nf1 blocked Braf-driven oncogene-induced senescence. Nf1-mutant and Braf-mutant tumours are resistant to BRAF inhibitors; however, they are sensitive to combined inhibition of MEK and mTOR162. Loss of NF1 was also identified as a crucial mediator of resistance to BRAF inhibitors in melanoma cells163,164. These studies indicate that blocking NF1 pathways — for example, by targeting MEK — might enhance the therapeutic response in those tumours with NF1 mutations. There are probably also other resistance pathways downstream of NF1. In a neuroblastoma model, resistance was mediated by NF1 through ZNF423, which encodes a zinc-finger transcription factor58.
It is increasingly clear that the recruitment of mast cells, macrophages and other stromal cells in the tumour microenvironment can elicit tumour cell resistance to therapy165. NF1−/− Schwann cells upregulate major histocompatibility complex class II mRNA and protein levels, which may influence tumour–immune cell interactions166. Neurofibromas and MPNSTs contain blood-derived mast cells167 and macrophages168. The stroma in NF1 neurofibromas has been recently reviewed169. These haematopoietic cells have a crucial role in the formation and growth of neurofibromas. In some GEM models, the haematopoietic cells must express mutant Nf1 for neurofibromas to form119. Treatment with PLX3397 — which targets both KIT signalling to prevent mast cell recruitment to tumours, and CSF1 receptor signalling to prevent macrophage recruitment to tumours — had two effects on neurofibromas. It increased their size when given during tumour initiation and enabled tumour regression in some mice when given after tumour establishment. Therefore, macrophages may protect against developing tumours and later become permissive to tumour formation168. However, wounding peripheral nerves in Nf1-mutant mice, which recruits macrophages, facilitates neurofibroma formation, implying that macrophages may promote tumour formation in this context83,170. OPGs that arise in Nf1-mutant mice also contain CX3CR1-expressing microglial cells (brain-resident macrophages). Cx3cr1−/− mice show delayed optic glioma formation, which supports interference with the micro-environment as a possible therapy in patients with NF1 (REF. 171). In an MPNST xenograft, PLX3397 treatment resulted in macrophage depletion and substantially delayed MPNST growth. The effect was enhanced by the mTORC1 inhibitor rapamycin and correlated with enhanced depletion of macrophages172.
Discovering new drug targets for NF1-mutant cells
Screening a library of 200,000 small molecules on mouse Nf1-mutant MPNST cells identified compound 21, with selectivity towards NF1-mutant cells and efficacy in xenografts173. Another library-screening study identified UC1 as a small molecule that targets NF1−/− versus sporadic MPNST cell lines. Budding yeast in which IRA2 was deleted validated selectivity of UC1 for NF1-mutant cells174. Direct targets of these compounds are not known. Gene expression, methylome and copy number changes on several sample sets are publically available for neurofibromas and MPNSTs, and are being used to identify pathways and targets for drug discovery175. Preliminary assessments of miRNAs and serum biomarkers have also become available. Several proteins were identified either at increased levels in patients with NF1 but without neurofibromas (interferon-γ, interleukin-6 and tumour necrosis factor) or at increased levels in patients with NF1 and MPNST (insulin-like growth factor-binding protein 1, C-C motif chemokine 5 (CCL5) and adrenomedullin)176,177. Expression of miR-801, miR-214 and miR-24 can distinguish patients with both NF1 and MPNST from patients with NF1 but without MPNST178. However, no NF1 biomarker has yet been tested clinically.
Successes and future challenges
The NF1 community has been very successful in identifying the NF1 gene and developing animal models for plexiform neurofibroma, MPNST and JMML. The community has also succeeded in identifying plausible therapeutic strategies and advancing them from preclinical testing to clinical trials, through preclinical and clinical testing consortia and a group developing end points for clinical trials179. However, some NF1-driven cancers still lack model systems or have models that are difficult to use for preclinical testing. Although the neurofibromin protein has been studied, many questions remain concerning the relevance of possible interaction partners and functions of neurofibromin protein domains. Although it is now clear that the RAS–MAPK pathway is crucial for mediating NF1-mutant tumour growth, other pathways downstream of RAS signalling are likely to be relevant and may be cell type dependent. In particular, an important goal of the next few years will be to better understand altered non-RAS–cAMP signalling downstream of NF1 loss. Identification of inhibitors of cell type-specific pathways that synergize with blockade of MEK, including inhibitors that target other RAS effector pathways, could be attempted. We anticipate that therapies successful in treating NF1 disease manifestations will also be successful in the treatment of other RASopathies and hope that these therapies, in combination with other drugs, will also be useful in treating sporadic NF1-mutant cancer.
Supplementary Material
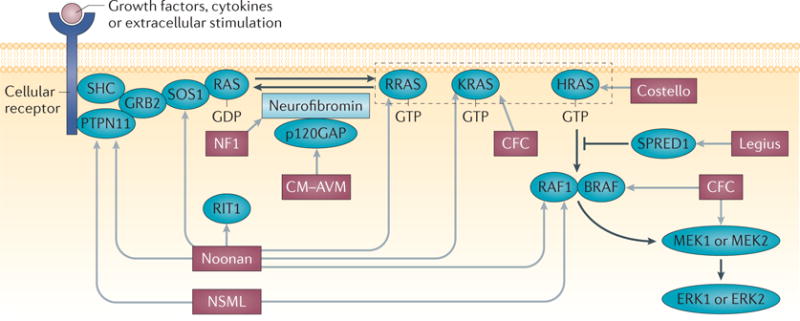
Box 1. RASopathy genes and syndromes.
RASopathies commonly predispose patients to short stature, developmental delay and cardiac abnormalities29. The syndrome (or syndromes) associated with each gene (or genes) is indicated by light grey arrows in the figure. RASopathies include the common disorders neurofibromatosis type 1 (NF1; which affects 1 in 3,000 individuals) and Noonan syndrome (which affects 1 in 2,000 individuals) and rare conditions, such as Costello syndrome. Mutations in several RAS–MAPK genes can cause Noonan syndrome, Noonan syndrome with multiple lentigines (NSML; previously known as LEOPARD syndrome) and cardio-facio-cutaneous (CFC) syndrome. Other RASopathies, including NF1, are associated with a single gene mutation. Legius syndrome is associated with mutations in sprouty-related, EVH1 domain-containing 1 (SPRED1), and capillary malformation–arteriovenous malformation (CM–AVM) is associated with mutations in RASA1 (which encodes p120GAP). RASopathy genes include protein tyrosine phosphatase non-receptor type 11 (PTPN11), CBL, son of sevenless homologue 1 (SOS1), RAF1, HRAS, NRAS, KRAS, BRAF, MAPK kinase 2 (MAP2K2) and SHOC2. New RASopathy genes continue to be identified; newly identified genes include RRAS180, RAS-like without CAAX 1 (RIT1)181, MAP2K1 and RASA2 (REF. 182). Many RASopathies predispose to tumours, including juvenile myelomonocytic leukaemia, rhabdomyosarcoma or neuroblastoma29. GRB2, growth factor receptor-bound protein 2; SHC, SRC homology 2 domain-containing.
Acknowledgments
The authors apologize to colleagues whose work they were unable to cite owing to space limitations or inadvertent omission. They have attempted to emphasize remaining questions and the most recent data in the field. They thank K. M. Cichowski (Brigham and Women’s Hospital, Massachusetts, USA), E. Schorry and N. Nassar (Cincinnati Children’s Hospital, Ohio, USA), and B. Widemann (US National Cancer Institute) for reviewing the draft manuscript. N.R. is supported by grants from the US National Institutes of Health, the Department of Defense Program on Neurofibromatosis, the Children’s Tumor Foundation and the Neurofibromatosis Therapeutic Acceleration Programs.
Glossary
- Café-au-lait macules
Hyperpigmented spots on the skin of patients with neurofibromatosis type 1 (NF1). They are used as an NF1 diagnostic criterion, particularly in young children
- Polycomb repressive complex 2
A complex that regulates epigenetic silencing of chromatin and includes the subunits SUZ12, EED, EZH1 or EZH2 and RBAP48. It also has histone methyltransferase activity
- Astrocytes
The most abundant type of glial cell in the central nervous system. Astrocytes regulate the extracellular neuronal environment
- Imprinting control region
A regulatory element (a segment of DNA) that is modified by methylation to regulate gene expression
- Schwann cells
Glial cells derived from neural crest cells that ensheathe and myelinate axons in the peripheral nervous system
- Oligodendrocytes
Glial cells derived from neuroepithelial cells that ensheathe and myelinate axons in the central nervous system
- NG2 cells
Oligodendrocyte progenitor cells that may have additional functions in the mature brain
- Aurora kinase
A serine/threonine kinase that functions during mitosis and is required for correct function of centrosomes
- Bromodomain inhibitors
A new class of epigenetic modulators of gene expression
Footnotes
Competing interests statement
The authors declare no competing interests.
FURTHER INFORMATION
cBio Portal for Cancer Genomics: http://www.cbioportal.org
Children’s Tumor Foundation: www.ctf.org
University of Alabama at Birmingham Medical Genomics Laboratory: http://www.uab.edu/medicine/genetics/medical-genomics-laboratory
SUPPLEMENTARY INFORMATION
See online article: S1 (table)
References
- 1.Crowe FW, Schull WJ, Neel JV. A Clinical, Pathological and Genetic Study of Multiple Neurofibromatosis. Charles C. Thomas; 1956. [Google Scholar]
- 2.Evans DG, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A:327–332. doi: 10.1002/ajmg.a.33139. [DOI] [PubMed] [Google Scholar]
- 3.Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989;26:704–711. doi: 10.1136/jmg.26.11.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman JM, Birch PH. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. Am J Med Genet. 1997;70:138–143. doi: 10.1002/(sici)1096-8628(19970516)70:2<138::aid-ajmg7>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 5.Huson SM, Compston DA, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. II. Guidelines for genetic counselling. J Med Genet. 1989;26:712–721. doi: 10.1136/jmg.26.11.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans DG, et al. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet. 2011;19:1187–1191. doi: 10.1038/ejhg.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125:63–66. doi: 10.1016/s0022-3476(94)70122-9. [DOI] [PubMed] [Google Scholar]
- 8.Neurofibromatosis Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575–578. [No authors listed.] [PubMed] [Google Scholar]
- 9.Gutmann DH, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57. [PubMed] [Google Scholar]
- 10.Stiller CA, Chessells JM, Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer. 1994;70:969–972. doi: 10.1038/bjc.1994.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walther MM, Herring J, Enquist E, Keiser HR, Linehan W. M von Recklinghausen’s disease and pheochromocytomas. J Urol. 1999;162:1582–1586. [PubMed] [Google Scholar]
- 12.Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–96. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 13.Stewart DR, et al. Diagnosis, management, and complications of glomus tumours of the digits in neurofibromatosis type 1. J Med Genet. 2010;47:525–532. doi: 10.1136/jmg.2009.073965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sung L, et al. Neurofibromatosis in children with rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Study IV. J Pediatr. 2004;144:666–668. doi: 10.1016/j.jpeds.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 15.Oktenli C, et al. Unusual features in a patient with neurofibromatosis type 1: multiple subcutaneous lipomas, a juvenile polyp in ascending colon, congenital intrahepatic portosystemic venous shunt, and horseshoe kidney. Am J Med Genet A. 2004;127A:298–301. doi: 10.1002/ajmg.a.30008. [DOI] [PubMed] [Google Scholar]
- 16.Viskochil D, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62:187–192. doi: 10.1016/0092-8674(90)90252-a. [DOI] [PubMed] [Google Scholar]
- 17.Cawthon RM, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 18.Wallace MR, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–186. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 19.Maertens O, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet. 2006;15:1015–1023. doi: 10.1093/hmg/ddl016. [DOI] [PubMed] [Google Scholar]
- 20.Brems H, et al. Glomus tumors in neurofibromatosis type 1: genetic, functional, and clinical evidence of a novel association. Cancer Res. 2009;69:7393–7401. doi: 10.1158/0008-5472.CAN-09-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madden JR, Rush SZ, Stence N, Foreman NK, Liu AK. Radiation-induced gliomas in 2 pediatric patients with neurofibromatosis type 1: case study and summary of the literature. J Pediatr Hematol Oncol. 2014;36:e105–e108. doi: 10.1097/MPH.0000000000000006. [DOI] [PubMed] [Google Scholar]
- 22.Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer. 2013;108:193–198. doi: 10.1038/bjc.2012.535. This paper suggests that individuals with NF1 are at risk of many types of cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballester R, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 24.Martin GA, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63:843–849. doi: 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- 25.Xu GF, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 26.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 27.Ohba Y, et al. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. J Biol Chem. 2000;275:20020–20026. doi: 10.1074/jbc.M000981200. This paper characterizes neurofibromin as an off signal for all RAS proteins. [DOI] [PubMed] [Google Scholar]
- 28.Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simsek-Kiper PO, et al. Clinical and molecular analysis of RASopathies in a group of Turkish patients. Clin Genet. 2013;83:181–186. doi: 10.1111/j.1399-0004.2012.01875.x. [DOI] [PubMed] [Google Scholar]
- 30.Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet. 2011;157C:83–89. doi: 10.1002/ajmg.c.30300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brems H, et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nature Genet. 2007;39:1120–1126. doi: 10.1038/ng2113. [DOI] [PubMed] [Google Scholar]
- 32.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meric-Bernstam F, et al. Concordance of genomic alterations between primary and recurrent breast cancer. Mol Cancer Ther. 2014;13:1382–1389. doi: 10.1158/1535-7163.MCT-13-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boudry-Labis E, et al. Neurofibromatosis-1 gene deletions and mutations in de novo adult acute myeloid leukemia. Am J Hematol. 2013;88:306–311. doi: 10.1002/ajh.23403. [DOI] [PubMed] [Google Scholar]
- 37.Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1) J Med Genet. 1996;33:2–17. doi: 10.1136/jmg.33.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Messiaen LM, et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15:541–555. doi: 10.1002/1098-1004(200006)15:6<541::AID-HUMU6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 39.Klose A, et al. Selective disactivation of neurofibromin GAP activity in neurofibromatosis type 1. Hum Mol Genet. 1998;7:1261–1268. doi: 10.1093/hmg/7.8.1261. This study identifies a point mutation that affects the neurofibromin RASGAP domain in a patient with NF1, which supports a key role for neurofibromin RASGAP activity in NF1. [DOI] [PubMed] [Google Scholar]
- 40.Fahsold R, et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet. 2000;66:790–818. doi: 10.1086/302809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balla B, et al. Fast and robust next-generation sequencing technique using ion torrent personal genome machine for the screening of neurofibromatosis type 1 (NF1) gene. J Mol Neurosci. 2014;53:204–210. doi: 10.1007/s12031-014-0286-7. [DOI] [PubMed] [Google Scholar]
- 42.Ko JM, Sohn YB, Jeong SY, Kim HJ, Messiaen LM. Mutation spectrum of NF1 and clinical characteristics in 78 Korean patients with neurofibromatosis type 1. Pediatr Neurol. 2013;48:447–453. doi: 10.1016/j.pediatrneurol.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Alkindy A, Chuzhanova N, Kini U, Cooper DN, Upadhyaya M. Genotype–phenotype associations in neurofibromatosis type 1 (NF1): an increased risk of tumor complications in patients with NF1 splice-site mutations? Hum Genomics. 2012;6:12. doi: 10.1186/1479-7364-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Raedt T, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003;72:1288–1292. doi: 10.1086/374821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Upadhyaya M, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970–2972 delAAT): evidence of a clinically significant NF1 genotype–phenotype correlation. Am J Hum Genet. 2007;80:140–151. doi: 10.1086/510781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rieley MB, et al. Variable expression of neurofibromatosis 1 in monozygotic twins. Am J Med Genet A. 2011;155A:478–485. doi: 10.1002/ajmg.a.33851. [DOI] [PubMed] [Google Scholar]
- 47.Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet. 1993;53:305–313. [PMC free article] [PubMed] [Google Scholar]
- 48.De Raedt T, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–251. doi: 10.1038/nature13561. [DOI] [PubMed] [Google Scholar]
- 49.Lee W, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nature Genet. 2014;46:1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang M, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nature Genet. 2014;46:1170–1172. doi: 10.1038/ng.3116. References 48–50 show that Polycomb repressive complex genes are commonly inactivated in MPNSTs and support sensitivity of MPNSTs to bromodomain-containing protein 4 inhibition in combination with MEK inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amlin-Van Schaick J, Kim S, Broman KW, Reilly KM. Scram 1 is a modifier of spinal cord resistance for astrocytoma on mouse chr 5. Mamm Genome. 2012;23:277–285. doi: 10.1007/s00335-011-9380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amlin-Van Schaick JC, et al. Arlm1 is a male-specific modifier of astrocytoma resistance on mouse chr 12. Neuro Oncol. 2012;14:160–174. doi: 10.1093/neuonc/nor206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saal HM, et al. Racial differences in the prevalence of optic nerve gliomas in neurofibromatosis type 1. Am J Hum Genet Abstr. 1995;57:A54. [Google Scholar]
- 54.Diggs-Andrews KA, et al. Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol. 2014;75:309–316. doi: 10.1002/ana.24093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun T, et al. Sexually dimorphic RB inactivation underlies mesenchymal glioblastoma prevalence in males. J Clin Invest. 2014;124:4123–4133. doi: 10.1172/JCI71048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ling JQ, et al. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312:269–272. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- 57.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holzel M, et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell. 2010;142:218–229. doi: 10.1016/j.cell.2010.06.004. This study is the first in a series of papers to demonstrate that NF1 mutation confers resistance to therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 60.Kim HA, Ling B, Ratner N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol Cell Biol. 1997;17:862–872. doi: 10.1128/mcb.17.2.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gutmann DH, et al. Heterozygosity for the neurofibromatosis 1 (NF1) tumor suppressor results in abnormalities in cell attachment, spreading and motility in astrocytes. Hum Mol Genet. 2001;10:3009–3016. doi: 10.1093/hmg/10.26.3009. [DOI] [PubMed] [Google Scholar]
- 62.Ingram DA, et al. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J Exp Med. 2000;191:181–188. doi: 10.1084/jem.191.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McGillicuddy LT, et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell. 2009;16:44–54. doi: 10.1016/j.ccr.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bajenaru ML, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–8577. [PubMed] [Google Scholar]
- 65.Zhu Y, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Daston MM, et al. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron. 1992;8:415–428. doi: 10.1016/0896-6273(92)90270-n. [DOI] [PubMed] [Google Scholar]
- 68.Hinman MN, Sharma A, Luo G, Lou H. Neurofibromatosis type 1 alternative splicing is a key regulator of Ras signaling in neurons. Mol Cell Biol. 2014;34:2188–2197. doi: 10.1128/MCB.00019-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bollag G, McCormick F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature. 1991;351:576–579. doi: 10.1038/351576a0. [DOI] [PubMed] [Google Scholar]
- 70.Vallee B, et al. Nf1 RasGAP inhibition of LIMK2 mediates a new cross-talk between Ras and Rho pathways. PLoS ONE. 2012;7:e47283. doi: 10.1371/journal.pone.0047283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stowe IB, et al. A shared molecular mechanism underlies the human rasopathies Legius syndrome and neurofibromatosis-1. Genes Dev. 2012;26:1421–1426. doi: 10.1101/gad.190876.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oliveira AF, Yasuda R. Neurofibromin is the major Ras inactivator in dendritic spines. J Neurosci. 2014;34:776–783. doi: 10.1523/JNEUROSCI.3096-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang HF, et al. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J Clin Invest. 2011;121:4820–4837. doi: 10.1172/JCI45677. References 71–73 provide the first strong evidence that specific neurofibromin-interacting proteins — SPRED1 and VCP — are crucial for neurofibromin function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tokuo H, et al. Phosphorylation of neurofibromin by cAMP-dependent protein kinase is regulated via a cellular association of NG,NG-dimethylarginine dimethylaminohydrolase. FEBS Lett. 2001;494:48–53. doi: 10.1016/s0014-5793(01)02309-2. [DOI] [PubMed] [Google Scholar]
- 75.Zhang P, et al. DDAH1 deficiency attenuates endothelial cell cycle progression and angiogenesis. PLoS ONE. 2013;8:e79444. doi: 10.1371/journal.pone.0079444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tan M, et al. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev Cell. 2011;21:1062–1076. doi: 10.1016/j.devcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hollstein PE, Cichowski K. Identifying the ubiquitin ligase complex that regulates the NF1 tumor suppressor and Ras. Cancer Discov. 2013;3:880–893. doi: 10.1158/2159-8290.CD-13-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rodenhiser DI, Andrews JD, Mancini DN, Jung JH, Singh SM. Homonucleotide tracts, short repeats and CpG/CpNpG motifs are frequent sites for heterogeneous mutations in the neurofibromatosis type 1 (NF1) tumour-suppressor gene. Mutat Res. 1997;373:185–195. doi: 10.1016/s0027-5107(96)00171-6. [DOI] [PubMed] [Google Scholar]
- 79.Gutmann DH, et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2013;23:431–439. doi: 10.1101/gr.142604.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lenarduzzi M, et al. MicroRNA-193b enhances tumor progression via down regulation of neurofibromin 1. PLoS ONE. 2013;8:e53765. doi: 10.1371/journal.pone.0053765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paschou M, Doxakis E. Neurofibromin 1 is a miRNA target in neurons. PLoS ONE. 2012;7:e46773. doi: 10.1371/journal.pone.0046773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Birnbaum RA, et al. Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol Cell. 2000;5:189–195. doi: 10.1016/s1097-2765(00)80415-3. [DOI] [PubMed] [Google Scholar]
- 83.Rizvi TA, et al. A novel cytokine pathway suppresses glial cell melanogenesis after injury to adult nerve. J Neurosci. 2002;22:9831–9840. doi: 10.1523/JNEUROSCI.22-22-09831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gouzi JY, et al. The receptor tyrosine kinase Alk controls neurofibromin functions in Drosophila growth and learning. PLoS Genet. 2011;7:e1002281. doi: 10.1371/journal.pgen.1002281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bollag G, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nature Genet. 1996;12:144–148. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 86.Jessen WJ, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340–347. doi: 10.1172/JCI60578. This study shows that, in a mouse model, inhibition of MEK shrinks neurofibromas, which supports ongoing clinical trials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dai C, et al. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest. 2012;122:3742–3754. doi: 10.1172/JCI62727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brundage ME, et al. MAF mediates crosstalk between Ras–MAPK and mTOR signaling in NF1. Oncogene. 2014;33:5626–5636. doi: 10.1038/onc.2013.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bodempudi V, et al. Ral overactivation in malignant peripheral nerve sheath tumors. Mol Cell Biol. 2009;29:3964–3974. doi: 10.1128/MCB.01153-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755–2760. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- 91.Johannessen CM, et al. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 92.Patmore DM, et al. In vivo regulation of TGFβ by R-Ras2 revealed through loss of the RasGAP protein Nf1. Cancer Res. 2012;72:5317–5327. doi: 10.1158/0008-5472.CAN-12-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Keng VW, et al. PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res. 2012;72:3405–3413. doi: 10.1158/0008-5472.CAN-11-4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Farrer RG, Farrer JR, DeVries GH. Platelet-derived growth factor-BB activates calcium/calmodulin-dependent and -independent mechanisms that mediate Akt phosphorylation in the neurofibromin-deficient human Schwann cell line ST88-14. J Biol Chem. 2013;288:11066–11073. doi: 10.1074/jbc.M112.442244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tanaka K, Matsumoto K, Toh E. A IRA1, an inhibitory regulator of the RAS–cyclic AMP pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:757–768. doi: 10.1128/mcb.9.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tanaka K, et al. IRA2, a second gene of Saccharomyces cerevisiae that encodes a protein with a domain homologous to mammalian ras GTPase-activating protein. Mol Cell Biol. 1990;10:4303–4313. doi: 10.1128/mcb.10.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Colombo S, Paiardi C, Pardons K, Winderickx J, Martegani E. Evidence for adenylate cyclase as a scaffold protein for Ras2–Ira interaction in Saccharomyces cerevisie. Cell Signal. 2014;26:1147–1154. doi: 10.1016/j.cellsig.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 98.Hegedus B, et al. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell. 2007;1:443–457. doi: 10.1016/j.stem.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 99.Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nature Neurosci. 2002;5:95–96. doi: 10.1038/nn792. [DOI] [PubMed] [Google Scholar]
- 100.Wolman MA, et al. Modulation of cAMP and Ras signaling pathways improves distinct behavioral deficits in a zebrafish model of neurofibromatosis type 1. Cell Rep. 2014;5:1265–1270. doi: 10.1016/j.celrep.2014.07.054. This recent study demonstrates that RAS and cAMP signalling, both altered by loss of neurofibromin function, are crucial for specific behavioural deficits in a model system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim HA, Ratner N, Roberts TM, Stiles CD. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J Neurosci. 2001;21:1110–1116. doi: 10.1523/JNEUROSCI.21-04-01110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anastasaki C, Gutmann DH. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum Mol Genet. 2014;23:6712–6721. doi: 10.1093/hmg/ddu389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tan X, et al. The CREB–miR-9 negative feedback minicircuitry coordinates the migration and proliferation of glioma cells. PLoS ONE. 2012;7:e49570. doi: 10.1371/journal.pone.0049570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Warrington NM, et al. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res. 2010;70:5717–5727. doi: 10.1158/0008-5472.CAN-09-3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rodriguez FJ, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008;67:240–249. doi: 10.1097/NEN.0b013e318165eb75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jones DT, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nature Genet. 2013;45:927–932. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhu Y, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132:5577–5588. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee YD, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22:131–138. doi: 10.1016/j.ccr.2012.05.036. This important paper suggests that the restricted localization of benign OPGs in children with NF1 occurs owing to the loss of NF1 in spatially restricted developing cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shin J, et al. Zebrafish neurofibromatosis type 1 genes have redundant functions in tumorigenesis and embryonic development. Dis Model Mech. 2012;5:881–894. doi: 10.1242/dmm.009779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mayes DA, et al. Nf1 loss and Ras hyperactivation in oligodendrocytes induce NOS-driven defects in myelin and vasculature. Cell Rep. 2013;4:1197–1212. doi: 10.1016/j.celrep.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Serra E, et al. Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell populations. Hum Mol Genet. 2000;9:3055–3064. doi: 10.1093/hmg/9.20.3055. [DOI] [PubMed] [Google Scholar]
- 112.Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453–463. doi: 10.1016/j.stem.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dugoff L, Sujansky E. Neurofibromatosis type 1 and pregnancy. Am J Med Genet. 1996;66:7–10. doi: 10.1002/(SICI)1096-8628(19961202)66:1<7::AID-AJMG2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 114.Prada CE, et al. Pediatric plexiform neurofibromas: impact on morbidity and mortality in neurofibromatosis type 1. J Pediatr. 2012;160:461–467. doi: 10.1016/j.jpeds.2011.08.051. [DOI] [PubMed] [Google Scholar]
- 115.Wu J, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–116. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Le LQ, et al. Susceptible stages in Schwann cells for NF1-associated plexiform neurofibroma development. Cancer Res. 2011;71:4686–4695. doi: 10.1158/0008-5472.CAN-10-4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mayes DA, et al. Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Res. 2011;71:4675–4685. doi: 10.1158/0008-5472.CAN-10-4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zheng H, et al. Induction of abnormal proliferation by nonmyelinating Schwann cells triggers neurofibroma formation. Cancer Cell. 2008;13:117–128. doi: 10.1016/j.ccr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 119.Yang FC, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/−- and c-kit-dependent bone marrow. Cell. 2008;135:437–448. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen Z, et al. Cells of origin in the embryonic nerve roots for NF1-associated plexiform neurofibroma. Cancer Cell. 2014;26:695–706. doi: 10.1016/j.ccell.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Williams JP, et al. Nf1 mutation expands an EGFR-dependent peripheral nerve progenitor that confers neurofibroma tumorigenic potential. Cell Stem Cell. 2008;3:658–669. doi: 10.1016/j.stem.2008.10.003. References 115–121 indicate that the neurofibroma cell of origin remains uncertain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Muir D, Neubauer D, Lim IT, Yachnis AT, Wallace MR. Tumorigenic properties of neurofibromin-deficient neurofibroma Schwann cells. Am J Pathol. 2001;158:501–513. doi: 10.1016/S0002-9440(10)63992-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tucker T, Wolkenstein P, Revuz J, Zeller J, Friedman JM. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005;65:205–211. doi: 10.1212/01.wnl.0000168830.79997.13. [DOI] [PubMed] [Google Scholar]
- 124.Carli M, et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol. 2005;23:8422–8430. doi: 10.1200/JCO.2005.01.4886. [DOI] [PubMed] [Google Scholar]
- 125.Evans DG, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–314. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Perrone F, et al. PDGFRA, PDGFRB, EGFR, and downstream signaling activation in malignant peripheral nerve sheath tumor. Neuro Oncol. 2009;11:725–736. doi: 10.1215/15228517-2009-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bottillo I, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693–701. doi: 10.1002/path.2494. [DOI] [PubMed] [Google Scholar]
- 128.Miller SJ, et al. Large-scale molecular comparison of human Schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 2006;66:2584–2591. doi: 10.1158/0008-5472.CAN-05-3330. [DOI] [PubMed] [Google Scholar]
- 129.Beert E, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–1032. doi: 10.1002/gcc.20921. This is a key paper demonstrating that mutations in CDKN2A are the only common chromosomal changes, apart from NF1 mutations, that are consistently detectable by single-nucleotide polymorphism analysis during plexiform neurofibroma transition to MPNST. [DOI] [PubMed] [Google Scholar]
- 130.Legius E, et al. TP53 mutations are frequent in malignant NF1 tumors. Genes Chromosomes Cancer. 1994;10:250–255. doi: 10.1002/gcc.2870100405. [DOI] [PubMed] [Google Scholar]
- 131.Menon AG, et al. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinhausen neurofibromatosis. Proc Natl Acad Sci USA. 1990;87:5435–5439. doi: 10.1073/pnas.87.14.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Birindelli S, et al. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest. 2001;81:833–844. doi: 10.1038/labinvest.3780293. [DOI] [PubMed] [Google Scholar]
- 133.De Raedt T, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for Ras-driven tumors. Cancer Cell. 2011;20:400–413. doi: 10.1016/j.ccr.2011.08.014. This is the first paper to identify a combination of therapies that target MPNSTs. The authors show that increasing proteotoxic stress causes tumour regression but only if combined with inhibition of the mTOR pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mawrin C, et al. Immunohistochemical and molecular analysis of p53, RB, and PTEN in malignant peripheral nerve sheath tumors. Virchows Arch. 2002;440:610–615. doi: 10.1007/s00428-001-0550-4. [DOI] [PubMed] [Google Scholar]
- 135.Mantripragada KK, et al. High-resolution DNA copy number profiling of malignant peripheral nerve sheath tumors using targeted microarray-based comparative genomic hybridization. Clin Cancer Res. 2008;14:1015–1024. doi: 10.1158/1078-0432.CCR-07-1305. [DOI] [PubMed] [Google Scholar]
- 136.Haferlach C, et al. AML with CBFB–MYH11 rearrangement demonstrate RAS pathway alterations in 92% of all cases including a high frequency of NF1 deletions. Leukemia. 2010;24:1065–1069. doi: 10.1038/leu.2010.22. [DOI] [PubMed] [Google Scholar]
- 137.Holtkamp N, et al. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008;10:946–957. doi: 10.1215/15228517-2008-053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Borrego-Diaz E, et al. Overactivation of Ras signaling pathway in CD133+ MPNST cells. J Neurooncol. 2012;108:423–434. doi: 10.1007/s11060-012-0852-1. [DOI] [PubMed] [Google Scholar]
- 139.Spyra M, et al. Cancer stem cell-like cells derived from malignant peripheral nerve sheath tumors. PLoS ONE. 2011;6:e21099. doi: 10.1371/journal.pone.0021099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cichowski K, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 141.Vogel KS, et al. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–2179. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dodd RD, et al. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol Cancer Ther. 2013;12:1906–1917. doi: 10.1158/1535-7163.MCT-13-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Joseph NM, et al. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell. 2008;13:129–140. doi: 10.1016/j.ccr.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Buchstaller J, McKeever PE, Morrison SJ. Tumorigenic cells are common in mouse MPNSTs but their frequency depends upon tumor genotype and assay conditions. Cancer Cell. 2012;21:240–252. doi: 10.1016/j.ccr.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang G, et al. Comparative oncogenomic analysis of copy number alterations in human and zebrafish tumors enables cancer driver discovery. PLoS Genet. 2013;9:e1003734. doi: 10.1371/journal.pgen.1003734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rahrmann EP, et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nature Genet. 2013;45:756–766. doi: 10.1038/ng.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.MacInnes AW, Amsterdam A, Whittaker CA, Hopkins N, Lees JA. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc Natl Acad Sci USA. 2008;105:10408–10413. doi: 10.1073/pnas.0805036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kumar MG, Emnett RJ, Bayliss SJ, Gutmann DH. Glomus tumors in individuals with neurofibromatosis type 1. J Am Acad Dermatol. 2014;71:44–48. doi: 10.1016/j.jaad.2014.01.913. [DOI] [PubMed] [Google Scholar]
- 149.Jacks T, et al. Tumor predisposition in mice heterozygous for a targeted mutation in NF1. Nature Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 150.Yoshimi A, Kojima S, Hirano N. Juvenile myelomonocytic leukemia: epidemiology, etiopathogenesis, diagnosis, and management considerations. Paediatr Drugs. 2010;12:11–21. doi: 10.2165/11316200-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 151.Chang T, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123:335–339. doi: 10.1172/JCI63193. This paper shows that, in a GEM JMML model, single-agent MEK inhibition significantly reduces disease, laying the foundation for human clinical trials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Robertson KA, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. Lancet Oncol. 2012;13:1218–1224. doi: 10.1016/S1470-2045(12)70414-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Marcus L, et al. Phase I study of the MEK1/2 inhibitor selumetinib (AZD6244) hydrogen sulfate in children and young adults with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PNs) J Clin Oncol Abstr. 2014;32:10018. [Google Scholar]
- 154.Katz D, Lazar A, Lev D. Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signalling pathways. Expert Rev Mol Med. 2009;11:e30. doi: 10.1017/S1462399409001227. [DOI] [PubMed] [Google Scholar]
- 155.Patel AJ, et al. BET bromodomain inhibition triggers apoptosis of NF1-associated malignant peripheral nerve sheath tumors through Bim induction. Cell Rep. 2014;6:81–92. doi: 10.1016/j.celrep.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Patel AV, et al. Ras-driven transcriptome analysis identifies aurora kinase A as a potential malignant peripheral nerve sheath tumor therapeutic target. Clin Cancer Res. 2012;18:5020–5030. doi: 10.1158/1078-0432.CCR-12-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mo W, et al. CXCR4/CXCL12 mediate autocrine cell cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell. 2013;152:1077–1090. doi: 10.1016/j.cell.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Watson AL, et al. Canonical Wnt/β-catenin signaling drives human Schwann cell transformation, progression, and tumor maintenance. Cancer Discov. 2013;3:674–689. doi: 10.1158/2159-8290.CD-13-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Luscan A, et al. The activation of the WNT signaling pathway is a hallmark in neurofibromatosis type 1 tumorigenesis. Clin Cancer Res. 2014;20:358–371. doi: 10.1158/1078-0432.CCR-13-0780. References 157–159 show the activation of WNT signalling in human MPNSTs and provide preclinical evidence that supports the importance of WNT signalling in MPNSTs. [DOI] [PubMed] [Google Scholar]
- 160.Kolberg M, et al. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol. 2013;15:135–147. doi: 10.1093/neuonc/nos287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.de Bruin EC, et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014;4:606–619. doi: 10.1158/2159-8290.CD-13-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Maertens O, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3:338–349. doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nissan MH, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74:2340–2350. doi: 10.1158/0008-5472.CAN-13-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Whittaker SR, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3:350–362. doi: 10.1158/2159-8290.CD-12-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. References 161–165 show that NF1 mutation causes resistance to therapy through multiple mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Reuss DE, et al. Functional MHC class II is upregulated in neurofibromin-deficient Schwann cells. J Invest Dermatol. 2013;133:1372–1375. doi: 10.1038/jid.2012.488. [DOI] [PubMed] [Google Scholar]
- 167.Carr NJ, Warren AY. Mast cell numbers in melanocytic naevi and cutaneous neurofibromas. J Clin Pathol. 1993;46:86–87. doi: 10.1136/jcp.46.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Prada CE, et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol. 2013;125:159–168. doi: 10.1007/s00401-012-1056-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yang FC, Staser K, Clapp DW. The plexiform neurofibroma microenvironment. Cancer Microenviron. 2012;5:307–310. doi: 10.1007/s12307-012-0115-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ribeiro S, et al. Injury signals cooperate with Nf1 loss to relieve the tumor-suppressive environment of adult peripheral nerve. Cell Rep. 2013;5:126–136. doi: 10.1016/j.celrep.2013.08.033. [DOI] [PubMed] [Google Scholar]
- 171.Pong WW, Higer SB, Gianino SM, Emnett RJ, Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol. 2013;73:303–308. doi: 10.1002/ana.23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Patwardhan PP, et al. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res. 2014;20:3146–3158. doi: 10.1158/1078-0432.CCR-13-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chau V, et al. Preclinical therapeutic efficacy of a novel pharmacologic inducer of apoptosis in malignant peripheral nerve sheath tumors. Cancer Res. 2014;74:586–597. doi: 10.1158/0008-5472.CAN-13-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wood M, et al. Discovery of a small molecule targeting IRA2 deletion in budding yeast and neurofibromin loss in malignant peripheral nerve sheath tumor cells. Mol Cancer Ther. 2011;10:1740–1750. doi: 10.1158/1535-7163.MCT-11-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Peacock JD, et al. Molecular-guided therapy predictions reveal drug resistance phenotypes and treatment alternatives in malignant peripheral nerve sheath tumors. J Transl Med. 2013;11:213. doi: 10.1186/1479-5876-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hummel TR, et al. Gene expression analysis identifies potential biomarkers of neurofibromatosis type 1 including adrenomedullin. Clin Cancer Res. 2010;16:5048–5057. doi: 10.1158/1078-0432.CCR-10-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Park SJ, et al. Serum biomarkers for neurofibromatosis type 1 and early detection of malignant peripheral nerve-sheath tumors. BMC Med. 2013;11:109. doi: 10.1186/1741-7015-11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Weng Y, Chen Y, Chen J, Liu Y, Bao T. Identification of serum microRNAs in genome-wide serum microRNA expression profiles as novel noninvasive biomarkers for malignant peripheral nerve sheath tumor diagnosis. Med Oncol. 2013;30:531. doi: 10.1007/s12032-013-0531-x. [DOI] [PubMed] [Google Scholar]
- 179.Gutmann DH, Blakeley JO, Korf BR, Packer RJ. Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin Investig Drugs. 2013;22:443–462. doi: 10.1517/13543784.2013.772979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Flex E, et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet. 2014;23:4315–4327. doi: 10.1093/hmg/ddu148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Aoki Y, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93:173–180. doi: 10.1016/j.ajhg.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chen PC, et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci USA. 2014;111:11473–11478. doi: 10.1073/pnas.1324128111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.von Recklinghausen F. Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen. August Hirschwald; 1882. [PubMed] [Google Scholar]
- 184.Sorensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. N Engl J Med. 1986;314:1010–1015. doi: 10.1056/NEJM198604173141603. [DOI] [PubMed] [Google Scholar]
- 185.Riccardi VM. Mast-cell stabilization to decrease neurofibroma growth. Preliminary experience with ketotifen. Arch Dermatol. 1987;123:1011–1016. [PubMed] [Google Scholar]
- 186.Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111:1355–1381. doi: 10.1093/brain/111.6.1355. [DOI] [PubMed] [Google Scholar]
- 187.Buchberg AM, Cleveland LS, Jenkins NA, Copeland NG. Sequence homology shared by neurofibromatosis type-1 gene and IRA-1 and IRA-2 negative regulators of the RAS cyclic AMP pathway. Nature. 1990;347:291–294. doi: 10.1038/347291a0. [DOI] [PubMed] [Google Scholar]
- 188.Brannan CI, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8:1019–1029. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- 189.Guo HF, The I, Hannan F, Bernards A, Zhong Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science. 1997;276:795–798. doi: 10.1126/science.276.5313.795. [DOI] [PubMed] [Google Scholar]
- 190.The I, et al. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 191.Zhu Y, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nguyen R, et al. Plexiform neurofibromas in children with neurofibromatosis type 1: frequency and associated clinical deficits. J Pediatr. 2011;159:652–655e2. doi: 10.1016/j.jpeds.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 193.DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105:608–614. doi: 10.1542/peds.105.3.608. [DOI] [PubMed] [Google Scholar]
- 194.Stevenson DA, et al. Descriptive analysis of tibial pseudarthrosis in patients with neurofibromatosis 1. Am J Med Genet. 1999;84:413–419. doi: 10.1002/(sici)1096-8628(19990611)84:5<413::aid-ajmg5>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 195.Alivuotila L, et al. Speech characteristics in neurofibromatosis type 1. Am J Med Genet A. 2010;152A:42–51. doi: 10.1002/ajmg.a.33178. [DOI] [PubMed] [Google Scholar]
- 196.Feldmann R, Denecke J, Grenzebach M, Schuierer G, Weglage J. Neurofibromatosis type 1: motor and cognitive function and T2-weighted MRI hyperintensities. Neurology. 2003;61:1725–1728. doi: 10.1212/01.wnl.0000098881.95854.5f. [DOI] [PubMed] [Google Scholar]
- 197.Nicolin G, et al. Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer. 2009;53:1231–1237. doi: 10.1002/pbc.22198. [DOI] [PubMed] [Google Scholar]
- 198.Hyman SL, Shores EA, North KN. Learning disabilities in children with neurofibromatosis type 1: subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Dev Med Child Neurol. 2006;48:973–977. doi: 10.1017/S0012162206002131. [DOI] [PubMed] [Google Scholar]
- 199.Garg S, et al. Autism and other psychiatric comorbidity in neurofibromatosis type 1: evidence from a population-based study. Dev Med Child Neurol. 2013;55:139–145. doi: 10.1111/dmcn.12043. [DOI] [PubMed] [Google Scholar]
- 200.Friedman JM, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med. 2002;4:105–111. doi: 10.1097/00125817-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 201.Ferner RE, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44:81–88. doi: 10.1136/jmg.2006.045906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Aravind L, Neuwald AF, Ponting CP. Sec14p-like domains in NF1 and Dbl-like proteins indicate lipid regulation of Ras and Rho signaling. Curr Biol. 1999;9:R195–R197. doi: 10.1016/s0960-9822(99)80127-4. [DOI] [PubMed] [Google Scholar]
- 203.Welti S, Fraterman S, D’Angelo I, Wilm M, Scheffzek K. The sec14 homology module of neurofibromin binds cellular glycerophospholipids: mass spectrometry and structure of a lipid complex. J Mol Biol. 2007;366:551–562. doi: 10.1016/j.jmb.2006.11.055. This study shows that crystallization of the neurofibromin lipid-binding domain supports the relevance of lipid interaction to neurofibromin function. [DOI] [PubMed] [Google Scholar]
- 204.D’Angelo I, Welti S, Bonneau F, Scheffzek K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep. 2006;7:174–179. doi: 10.1038/sj.embor.7400602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.